Gözlemci-beklenti etkisi araştırmacı veya analizcinin kişisel çıkarları doğrultusunda bilinçsizce sonucu etkilemesi nedeniyle ortaya çıkan önyargıyı ifade eder.
Örnek: Bir doktor, miyokard enfarktüsü ve mide ağrısı öncesinde mide ağrısının oluşumu hakkında bir çalışma yapmayı planlamaktadır. Seçilen hastalar sadece doktorun doğrudan bu soruyu sormasının ardından mide ağrısına benzer bir his yaşadıklarını belirtmişlerdir. Eğer doktor bu soruyu sormamış olsaydı, katılımcıların hiçbiri kalp krizi ile birlikte mide ağrısı yaşadığını belirtmeyecekti. Doktorun sorusu hastaların olumlu yanıt vermesini sağlamış, bu da hastaların güvenilirliğini ve dolayısıyla mide ağrısının yaklaşan bir kalp krizinin göstergesi olabileceği hipotezini sorgulatmıştır.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Biyoistatistik veya Epidemiyolojide yanlılık, istenen sonucu üreten herhangi bir süreci etkileyen tahmin edilmemiş veya hesaplanmamış faktörler nedeniyle sonucun tahrif edilmesi anlamına gelir.
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Karıştırıcı yanlılık, maruziyet ve sonuç arasındaki ilişkiyi etkileyen hesapa katılmamış bir faktör mevcut olduğunu tarifler. Ya yokluğunda olmayacak bir ilişki oluşturur ya da bir ilişkiyi maskeler. Her iki durumda da, kohort çalışmalarında görüldüğü gibi maruziyete bağlı bir çalışmanın sonucu değişebilir.
Örnek. Bir çalışmada, A ilacının kan glikozunu düşürme etkisi test edilmiştir. A ilacını alacak olan grupta, bu ilacın metabolizmasına müdahale eden ve böylece etkisini azaltan bir hastalık için farklı ilaç alan daha fazla birey vardır. Bu durum, bazı bireylerde ilacın gerçek etkisini maskeleyerek sonuçları değiştirebilir. Randomizasyon bu yanlılığın azaltılmasına yardımcı olmaktadı.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Teslim süresi, bir hastalığın tarama yoluyla tespit edildiği andan, söz konusu hastalığın herhangi bir tarama yapılmaksızın klinik ifadesi nedeniyle teşhis edildiği ana kadar geçen süreyi ifade eder. Hastalığın seyri üzerinde herhangi bir etkisi olmaksızın sağkalım süresine yanlışlıkla dahil edilmesi durumunda, Teslim süresi sapması meydana gelir.
Örnek. A hastalığı olarak adlandırılan ve oldukça spesifik bir testle teşhis edilen bir hastalık olduğunu varsayalım. Herhangi bir klinik bulgu olmaksızın hekimi A hastalığı olasılığı konusunda uyarabilen yeni bir tarama testi uygulamaya konmuştur. Şimdi ileriye dönük bir kohort çalışması başlatıldı. Deney ya da kontrol grubu olmak üzere her iki gruptaki tüm bireyler, ileri yaşları ve dolayısıyla tedaviye uygun olmamaları nedeniyle tedavi görmemektedir.
Çalışmadan veriler toplanmıştır. Araştırmacılar, tarama yapılan deney grubundaki kişilerin, tarama yapılmayan bireylere göre daha yüksek bir sağkalım oranına sahip olduğunu kabul etselerdi, bir öncü zaman yanlılığı ortaya çıkacaktı. Tarama yapılan kişilere pozitif tarama nedeniyle daha erken tanı konulduğundan, tanı konulduğu andan ölüme kadar geçen sürede hayatta kalma oranları daha yüksektir. Tarama süresi hayatta kalma oranını etkilemez. Bu örnekte, yaşa özgü ölüm oranı da incelenmeliydi.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Kardiyak arrest, kalbin aniden etkili bir şekilde atmayı bırakması ve bunun sonucunda beyne ve diğer hayati organlara kan gitmemesi durumunda ortaya çıkan tıbbi bir acil durumdur (Ani Kalp Durması). Bu durum solunumun durmasına ve bilinç kaybına yol açar. Kardiyak arrest, fonksiyonun ve normal kalp ritminin yeniden sağlanması için acil müdahale gerektiren, yaşamı tehdit eden bir olaydır.
Belirtiler ve bulgular:
Ani bilinç kaybı: Kişi uyarı vermeden yere yığılır ve bilincini kaybeder.
Devre yok: Palpe edilebilir nabız veya tespit edilebilir kan basıncı yoktu.
Solunum yok veya anormal solunum: Solunum tamamen durabilir veya soluk soluğa ve düzensiz olabilir.
Soluk ve soluk cilt: Dolaşımdaki oksijen eksikliği nedeniyle cilt soluklaşabilir veya solgunlaşabilir.
Yanıt yok: Kişi dış uyaranlara yanıt vermez.
Teşhis koyun:
Kardiyak arrest tanısı ani bilinç kaybı, nabız kaybı ve solunumun durmasına dayanır. Acil müdahale gereklidir ve tanısal prosedürler genellikle resüsitasyon çabaları başladıktan sonra gerçekleştirilir.
Tedavi:
Kardiyak arrest acil müdahale gerektiren tıbbi bir acil durumdur:
Yardım çağırın: Hemen acil servisleri arayın veya yerel acil müdahale sisteminizi etkinleştirin.
Kardiyopulmoner resüsitasyonu (CPR)başlatın: Kan dolaşımına yardımcı olmak ve hayati organlara oksijen sağlamak için göğüs kompresyonlarına başlayın. Profesyonel tıbbi yardım gelene kadar kalp masajı yapılmalıdır.
İleri Yaşam Desteği: Tıp uzmanları ilaçları, gelişmiş hava yolu yönetimini uygulayacak ve sabit bir kalp ritmini geri getirmeye çalışmak için gelişmiş kardiyak yaşam desteği tekniklerini (ACLS) kullanacaktır.
Altta yatan nedeni tedavi edin: Kişinin kalp atışı yeniden sağlandıktan sonra, sağlık ekibi kalp durmasının altında yatan nedeni belirlemek ve tedavi etmek için çalışacaktır.
Ayırıcı tanı:
Kardiyak arrest, bilinç kaybına ve kollapsa yol açabilecek diğer durumlardan ayırt edilmelidir:
Bayılma: Bayılma, genellikle iyi huylu bir nedenden dolayı beyne giden kan akışında geçici bir azalmadan kaynaklanır.
Giriş: Beyindeki anormal elektriksel aktivite istemsiz hareketlere ve bilinç bozukluğuna yol açar.
Yarış: Beyne giden kan akışının aniden kesilmesi nörolojik semptomlara neden olur.
Aşırı dozda ilaç kullanımı: Bazı ilaçlar bilinç kaybına ve anormal hayati belirtilere neden olabilir.
Hipoglisemi: Aşırı düşük kan şekeri zihinsel durumun değişmesine ve çöküşe yol açabilir.
Şok: Vücudun organlarının yeterli kan ve oksijen alamadığı, yaşamı tehdit eden bir durumdur. Kalp durmasının ciddi bir tıbbi acil durum olduğunu ve acil müdahale gerektirdiğini unutmamak önemlidir. Kalp masajı, defibrilasyon ve ileri tıbbi müdahalenin hemen başlatılması hayatta kalma şansını büyük ölçüde artırır.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Taşikardi, genellikle yetişkinlerde dakikada 100 atımdan (bpm) daha yüksek bir dinlenme kalp atış hızı olarak tanımlanan anormal derecede hızlı bir kalp atış hızını ifade eder. Bazı durumlarda taşikardi egzersiz, stres veya ateşe verilen normal bir fizyolojik yanıt olabilir. Bununla birlikte, inatçı veya tekrarlayan taşikardi altta yatan tıbbi bir duruma işaret edebilir.
Belirtiler ve bulgular:
Utanmış: Hızlı, çarpan veya düzensiz kalp atışı hissi.
Baş dönmesi veya sersemlik: Hızlı kalp atışı nedeniyle beyne giden kan akışının azalması.
Nefes almada güçlük: Kalp etkili bir şekilde pompalayamadığı için vücuda yeterli oksijen gitmemesi.
Rahatsızlık veya göğüs ağrısı: Genellikle göğüste baskı veya rahatsızlık olarak tanımlanır.
Yorgunluk: Kalbin iş yükünün artması ve verimliliğinin azalması nedeniyle.
Bayılma veya bayılmaya yakın olma: Özellikle taşikardi kan basıncında keskin bir düşüşe neden oluyorsa.
Teşhis edin:
Elektrokardiyogram (EKG/EKG): Kalbin elektriksel aktivitesini kaydetmek için yapılan standart bir testtir. Taşikardinin türünü ve altında yatan nedeni belirlemeye yardımcı olur.
Holter Ekranı: Sürekli kalp atış hızı takibi için hasta tarafından 24 ila 48 saat boyunca takılan taşınabilir bir EKG cihazı.
Olay izleme: Holter monitörüne benzer, ancak ara sıra taşikardi ataklarını kaydetmek için daha uzun süre takılır.
Ekokardiyografi: Ekokardiyogram, kalbin yapısı ve işlevinin ayrıntılı görüntülerini sağlar.
Kan testleri: Dengesizlik taşikardiye katkıda bulunabileceğinden elektrolit seviyelerini ve tiroid fonksiyonunu değerlendirmek için.
Tedavi:
Tedavi, taşikardinin altında yatan nedene ve semptomların şiddetine bağlıdır:
Vajinal prosedür: Öksürmek, bastırmak veya yüzünüzü soğuk suya daldırmak gibi basit teknikler vagus sinirini uyarır ve kalp atış hızını yavaşlatır.
İlaçlar: Kalp hızını ve ritmini kontrol etmek için antiaritmik ilaçlar reçete edilebilir.
Kalp Hızı Transferi: Acil bir durumda normal kalp ritmini yeniden sağlamak için elektrokonvülsif tedavi.
Kesim: Kalpteki anormal elektrik yollarını hedeflemek ve çıkarmak için kateter tabanlı prosedür.
Kalp pili: İmplante edilebilir cihaz, kalp atış hızını gerektiği gibi izler ve kontrol eder.
Yaşam tarzınızı değiştirin: Stresi azaltmak, kafein ve alkolü sınırlamak ve susuz kalmamak taşikardiyi kontrol etmeye yardımcı olabilir.
Ayırıcı tanı:
Benzer semptomlara neden olabilen ve taşikardiden ayırt edilmesi gereken diğer durumlar şunlardır:
Endişe: Stres veya endişe nedeniyle kalp atış hızının artması.
Ateş: Yüksek vücut sıcaklığına tepki olarak kalp atış hızı artar.
Güçlendirilmiş Zırh: Aşırı aktif bir tiroid metabolizma hızının ve kalp atış hızının artmasına neden olur.
Anemi: Kanın oksijen taşıma kapasitesinin azalması kalp atış hızının artmasına neden olur.
Kan hacminde azalma: Düşük kan hacmi, kan basıncını korumak için kalp atış hızının artmasına neden olur.
Kalp Yetmezliği: İlerleyen aşamalarda kalp, bozulmuş pompalama yeteneğini telafi etmek için daha hızlı atabilir.
Taşikardinin yönetimi ve tedavisinin spesifik tipe ve altta yatan nedene bağlı olarak değiştiğini unutmamak önemlidir. Bu nedenle, uygun yöntemi belirlemek için uygun tıbbi değerlendirme gereklidir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Taşikardi, genellikle yetişkinlerde dakikada 100 atımdan (bpm) daha yüksek bir dinlenme kalp atış hızı olarak tanımlanan anormal derecede hızlı bir kalp atış hızını ifade eder. Bazı durumlarda taşikardi egzersiz, stres veya ateşe verilen normal bir fizyolojik yanıt olabilir. Bununla birlikte, inatçı veya tekrarlayan taşikardi altta yatan tıbbi bir duruma işaret edebilir.
Belirtiler ve bulgular
Utanmış: Hızlı, çarpan veya düzensiz kalp atışı hissi.
Baş dönmesi veya sersemlik: Hızlı kalp atışı nedeniyle beyne giden kan akışının azalması.
Nefes almada güçlük: Kalp etkili bir şekilde pompalayamadığı için vücuda yeterli oksijen gitmemesi.
Rahatsızlık veya göğüs ağrısı: Genellikle göğüste baskı veya rahatsızlık olarak tanımlanır.
Yorgunluk: Kalbin iş yükünün artması ve verimliliğinin azalması nedeniyle.
Bayılma veya bayılmaya yakın olma: Özellikle taşikardi kan basıncında keskin bir düşüşe neden oluyorsa.
Teşhis edin:
Elektrokardiyogram (EKG/EKG): Kalbin elektriksel aktivitesini kaydetmek için yapılan standart bir testtir. Taşikardinin türünü ve altında yatan nedeni belirlemeye yardımcı olur.
Holter Ekranı: Sürekli kalp atış hızı takibi için hasta tarafından 24 ila 48 saat boyunca takılan taşınabilir bir EKG cihazı.
Olay izleme: Holter monitörüne benzer, ancak ara sıra taşikardi ataklarını kaydetmek için daha uzun süre takılır.
Ekokardiyografi: Ekokardiyogram, kalbin yapısı ve işlevinin ayrıntılı görüntülerini sağlar.
Kan testleri: Dengesizlik taşikardiye katkıda bulunabileceğinden elektrolit seviyelerini ve tiroid fonksiyonunu değerlendirmek için.
Tedavi:
Tedavi, taşikardinin altında yatan nedene ve semptomların şiddetine bağlıdır:
Vajinal prosedür: Öksürmek, bastırmak veya yüzünüzü soğuk suya daldırmak gibi basit teknikler vagus sinirini uyarır ve kalp atış hızını yavaşlatır.
İlaçlar: Kalp hızını ve ritmini kontrol etmek için antiaritmik ilaçlar reçete edilebilir.
Kalp Hızı Transferi: Acil bir durumda normal kalp ritmini yeniden sağlamak için elektrokonvülsif tedavi.
Kesim: Kalpteki anormal elektrik yollarını hedeflemek ve çıkarmak için kateter tabanlı prosedür.
Kalp pili: İmplante edilebilir cihaz, kalp atış hızını gerektiği gibi izler ve kontrol eder.
Yaşam tarzınızı değiştirin: Stresi azaltmak, kafein ve alkolü sınırlamak ve susuz kalmamak taşikardiyi kontrol etmeye yardımcı olabilir.
Ayırıcı tanı:
Benzer semptomlara neden olabilen ve taşikardiden ayırt edilmesi gereken diğer durumlar şunlardır:
Güçlendirilmiş Zırh: Aşırı aktif bir tiroid metabolizma hızının ve kalp atış hızının artmasına neden olur.
Anemi: Kanın oksijen taşıma kapasitesinin azalması kalp atış hızının artmasına neden olur.
Kan hacminde azalma: Düşük kan hacmi, kan basıncını korumak için kalp atış hızının artmasına neden olur.
Kalp Yetmezliği: İlerleyen aşamalarda kalp, bozulmuş pompalama yeteneğini telafi etmek için daha hızlı atabilir. Taşikardinin yönetimi ve tedavisinin spesifik tipe ve altta yatan nedene bağlı olarak değiştiğini unutmamak önemlidir. Bu nedenle, uygun yöntemi belirlemek için uygun tıbbi değerlendirme gereklidir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Pulmoner Emboli için Wells Kriterleri, bir hastanın akciğer atardamarında kan pıhtısı oluştuğu ve hayatı tehdit eden bir durum olan pulmoner emboli (PE) geliştirme olasılığını değerlendirmek için kullanılan klinik bir karar kuralıdır. Kriterler, sağlık çalışanlarının pulmoner emboli varlığını doğrulamak veya ekarte etmek için görüntüleme çalışmaları gibi diğer tanısal testlerin yapılıp yapılmayacağını belirlemelerine yardımcı olur. Wells kriterleri, pulmoner emboli ile ilişkili çeşitli klinik faktörleri ve semptomları dikkate alır.
Wells Kriterlerinin iki versiyonu vardır:
Orijinal versiyon ve basitleştirilmiş versiyon (Klinik Kuruluşta PE için Wells Kriterleri). Burada daha kapsamlı bir inceleme sunarak orijinal versiyonu açıklayacağım. Wells temel çizgisi aşağıdaki klinik faktörleri içerir ve her bir faktöre bir puan atar:
Derin ven trombozunun (DVT) klinik belirti ve semptomları: Bacaklarda DVT’nin klinik belirti ve semptomları (örn. şişme, ağrı): 3 puan.
Pulmoner emboliden daha az olası alternatif tanı: Daha az olası bir alternatif tanı: 3 puan.
Kalp atışı: Kalp atım hızı >100 atım/dak: 1,5 nokta.
Yakın zamanda gayrimenkul:Son 4 hafta içinde > 3 gün immobilizasyon veya büyük ameliyat: 1,5 nokta.
Derin ven trombozu veya pulmoner emboli öyküsü: Önceden DVT veya PE: 1,5 nokta.
Kan öksürme: Kan öksürme (kan tükürme):1 nokta.
Malignite: Kötü huylu hastalık (tedavi altında veya son 6 ay içinde): 1 puan.
Bu faktörler değerlendirilip puanlandıktan sonra toplam puan hesaplanır. Wells Kriterleri skorunun yorumlanması aşağıdaki gibidir:
Düşük olasılık (ölçek 6):
PE olasıdır ve ek tanı ve görüntüleme testleri önerilir. Wells kriterinin klinik karar vermeye rehberlik eden bir araç olduğunu ve kesin bir tanı olmadığını unutmamak önemlidir. Hastaları risk kategorilerine ayırmaya ve ileri test ihtiyacını belirlemeye yardımcı olur. Kan pıhtısı oluşumunun belirteçlerini ölçen D-dimer kan testi, PE olasılığını daha fazla değerlendirmek için genellikle Wells kriterleri ile birlikte kullanılır. Klinisyenler ileri değerlendirme ve tedavi hakkında karar verirken hastanın genel klinik tablosunu, tıbbi geçmişini ve diğer ilgili faktörleri de göz önünde bulundurmalıdır. Wells Kriterleri, kapsamlı bir pulmoner emboli değerlendirmesinin yalnızca bir bileşenidir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Cockcroft-Gault denklemi, bir kişinin böbrek fonksiyonunun bir ölçüsü olan kreatinin klirensini tahmin etmek için yaygın olarak kullanılan bir formüldür. Kreatinin, kaslar tarafından yapılan ve böbrekler tarafından atılan bir atık üründür. Kreatinin klirensi, böbreklerin kreatinini kandan ne kadar verimli bir şekilde uzaklaştırdığını yansıtır. Bu ölçüm, özellikle böbrek tarafından elimine edilen ilaçlar kullanıldığında böbrek fonksiyonunu değerlendirmek için önemlidir.
Cockcroft-Gault denklemi, kreatinin klirensini tahmin etmek için kişinin yaşını, kilosunu, cinsiyetini ve serum kreatinin seviyesini dikkate alır. Formül şöyledir:
Kreatinin klirensi (mL/dak) = [(140 – Yaş) × Ağırlık (kg)] / [72 × Serum kreatinin (mg/dL)]
Erkekler için 140 ve kadınlar için 85 sabittir. Cockcroft-Gault denkleminin özellikle belirli popülasyonlarda sınırlamaları olduğuna dikkat etmek önemlidir. Örneğin
Farklı yaş gruplarında doğruluk: Denklem, kas kütlesi ve kreatinin üretimindeki farklılıklar nedeniyle çok genç veya çok yaşlı bireylerde kreatinin klirensini doğru olarak tahmin edemeyebilir.
Vücut kompozisyonu: Denklem ortalama kas kütlesini varsayar, bu da çok farklı vücut kompozisyonlarına sahip kişilerde (örneğin sporcular, atrofi veya amputasyonu olan kişiler) yanlışlıklara neden olabilir.
Kararsız koşullar: Kreatinin üretiminin ve böbrek fonksiyonunun hızla değiştiği durumlarda (örn. akutböbrek hasarı) denklemin doğruluğu etkilenebilir.
Cinsiyet: Denklem, kas kütlesinde bir fark olduğunu varsayarak erkekler ve kadınlar için farklı bir sabit kullanır. Ancak, bu tüm bireyler için doğru olmayabilir.
Modern laboratuvar teknikleri ile doğruluk: Cockcroft-Gault denklemi, serum kreatininini ölçmek için eski laboratuvar yöntemleri kullanılarak geliştirilmiştir. Böbrek Hastalığında Modifiye Diyet Denklemi (MDRD) veya Ortak KronikBöbrek Hastalığı Epidemiyoloji Denklemi (CKD-EPI) gibi daha modern denklemler daha doğru tahminler için sıklıkla önerilmektedir.
Kısıtlamalarına rağmen Cockcroft-Gault denklemi, basitliği ve tarihsel önemi nedeniyle özellikle klinik uygulamada hala yaygın olarak kullanılmaktadır. Bununla birlikte, klinik bağlama ve mevcut hasta verilerine bağlı olarak, sağlık hizmeti sağlayıcıları, özellikle farklı popülasyonlarda böbrek fonksiyonu tahminlerinin doğruluğunu artırmak için geliştirilen daha modern denklemleri kullanmayı da düşünebilirler.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
4Ts Skoru, heparin tedavisine karşı potansiyel olarak ciddi bir immün aracılı advers reaksiyon olan Heparine Bağlı Trombositopeni (HIT) olasılığını değerlendirmek için kullanılan klinik bir skorlama sistemidir. HIT, trombosit sayısında önemli bir azalma ile karakterize edilir ve bu da kan pıhtısı oluşumu riskinin artmasına neden olabilir. 4Ts Skoru, klinisyenlerin dört temel klinik faktöre dayanarak HIT olasılığını değerlendirmelerine yardımcı olur: Trombositopeni, Zamanlama, Tromboz ve trombositopeninin diğer nedenleri.
İşte 4Ts Skoru bileşenlerinin ayrıntılı bir dökümü:
Trombositopeni:
2 puan: Trombosit sayısının ≥%50 düşmesi veya trombosit sayısının <150 x 10^9/L olması.
1 puan: Trombosit sayısı %30-49 oranında düşer.
Trombositopeninin Zamanlaması:
2 puan: Trombositopeni heparine başladıktan 5-10 gün sonra ortaya çıkar.
1 puan: Trombositopeni heparin başlandıktan sonra 1-4 gün içinde veya >10 gün içinde ortaya çıkar.
Tromboz veya diğer sekeller:
2 puan: Tromboz (yeni veya ilerleyici) veya cilt nekrozu oluşur.
1 puan: Anafilaktoid reaksiyon veya tromboz olmaksızın şüpheli HIT gibi akut sistemik reaksiyon.
Trombositopeninin diğer nedenleri:
2 puan: Trombositopeni için başka bir açıklama yok.
1 puan: Olası diğer nedenler mevcuttur.
Toplam puan, dört bileşenin her birine verilen puanların toplanmasıyla elde edilir. 4Ts Skorunun genel yorumu aşağıdaki gibidir:
Yüksek Olasılık (6-8 puan): HIT olasılığı yüksektir ve derhal ileri değerlendirme ve yönetim uygulanmalıdır.
Orta Olasılık (4-5 puan): HIT olasılığı orta düzeydedir. Klinik değerlendirme ve ek testler gerekebilir.
Düşük Olasılık (0-3 puan): HIT olasılığı düşüktür. Trombositopeninin alternatif nedenleri araştırılmalıdır ve HIT için daha fazla araştırma gerekli olmayabilir.
4Ts Skorunun klinik bir değerlendirme aracı olduğunu ve kesin tanı kararları vermek için tek başına kullanılmaması gerektiğini unutmamak önemlidir. Genellikle tanıyı doğrulamak için HIT antikor testi gibi ek laboratuvar testlerinin gerekli olup olmadığını belirlemek için bir kılavuz olarak kullanılır. HIT riski değerlendirilirken diğer klinik faktörler, hasta geçmişi ve tıbbi bağlam da göz önünde bulundurulur.
4Ts Skoru, HIT olasılığını değerlendirmek için yapılandırılmış bir yaklaşım sunarak sağlık hizmeti sağlayıcılarının heparin tedavisi sırasında şüpheli veya doğrulanmış trombositopenisi olan hastalarda heparin kullanımı ve potansiyel alternatifler konusunda bilinçli kararlar almasına yardımcı olur.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Ulusal Sağlık Enstitüleri İnme Ölçeği (NIHSS), inme şiddetini değerlendirmek ve hastaların nörolojik durumunu izlemek için yaygın olarak kullanılan bir klinik araçtır. Ulusal Nörolojik Bozukluklar ve İnme Enstitüsü (NINDS) tarafından inmeye bağlı nörolojik defisitleri ölçmenin standart bir yolu olarak geliştirilmiştir. Ölçek, sağlık çalışanları tarafından, özellikle inme ve acil bakım ortamlarında, inme nedeniyle hastaların nörolojik bozulma derecesini değerlendirmek ve iletmek için kullanılır.
NIHSS, her biri nörolojik fonksiyonun belirli bir yönünü değerlendiren 11 maddeden oluşmaktadır. Maddeler bilinç, motor beceriler, duyusal algı, konuşma ve dil dahil olmak üzere çok çeşitli işlevleri ölçmektedir. Gözlemlenen eksikliklerin ciddiyetine göre her bir maddeye puan verilir. Her bir maddenin puanlarının toplanmasıyla elde edilen toplam puan, 0 (eksiklik yok) ile maksimum 42 puan (nöral eksiklikler) arasında değişen genel bir inme şiddeti değerlendirmesi sağlar. ciddi).
İlk olarak: Görsel, dokunsal, işitsel, uzamsal veya kişisel dikkatsizlik veya birden fazla modaliteye dikkatsizlik Sağlık çalışanları NIHSS’yi hastaların nörolojik durumlarını başvuru anında ve sonrasında aralıklarla değerlendirmek ve durumlarındaki değişiklikleri izlemek için kullanmaktadır.
Skorlar tedavi kararlarını yönlendirmeye, sonuçları tahmin etmeye yardımcı olabilir ve inme müdahalesi ile ilgili araştırma ve klinik çalışmalar için değerli bilgiler sağlayabilir. NIHSS değerli bir araç olmakla birlikte, inme değerlendirmesinin yalnızca bir yönü olduğunu ve hastanın durumunun tam olarak anlaşılması için görüntüleme çalışmaları ve diğer klinik bilgilerin de dikkate alındığını belirtmek önemlidir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Standart sapma, bir dizi veri noktasındaki varyasyon veya dağılım miktarını ölçen istatistiksel bir ölçüdür. Bir veri setindeki belirsizlik veya yayılma miktarının yaygın olarak kullanılan bir ölçüsüdür. Daha küçük bir standart sapma, veri noktalarının ortalamaya (ortalamaya) yakın olma eğiliminde olduğunu gösterirken, daha büyük bir standart sapma veri noktalarının ortalamadan daha fazla dağıldığını gösterir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Nötropeni, kanda anormal derecede düşük nötrofil seviyeleri ile karakterize bir durumu ifade eden tıbbi bir terimdir. Nötrofiller bir tür beyaz kan hücresi (WBC) olup vücudun bağışıklık sisteminin önemli bir parçasıdır ve enfeksiyonlarla, özellikle de bakteriyel enfeksiyonlarla mücadelede önemli bir rol oynar. Nötrofil sayısı normalden düşük olduğunda, enfeksiyon ve diğer sağlık komplikasyonları riskinin artmasına neden olabilir.
Nötropeni genellikle mutlak nötrofil sayısının (ANC) belirli bir eşiğin altında olması olarak tanımlanır. Kan nötrofillerinin normal aralığı genellikle mikrolitre (μL) kan başına 1.500 ila 8.000 nötrofil arasındadır. Nötropeni, ANC’ye bağlı olarak çeşitli tiplerde sınıflandırılır:
Hafif lökopeni: ANC 1000 ila 1500/μL aralığındadır.
Orta derecede lökopeni: ANC 500 ila 1000/μL aralığında.
** Şiddetli nötropeni**: ANC değerinin 500/μL’den az olması. Nötropeni birçok faktörden kaynaklanabilir ve altta yatan neden ciddiyetini ve tedavisini belirler. Nötropeninin bazı yaygın nedenleri şunlardır:
Enfeksiyon: Hepatit, HIV ve influenza gibi bazı viral enfeksiyonlar geçici nötropeniye neden olabilir.
İlaç : Bazı ilaçlar, özellikle bazı kemoterapi ilaçları, yan etki olarak nötropeniye neden olabilir. Bazı antibiyotikler veya nöbet önleyici ilaçlar gibi diğer ilaçlar da nötrofil seviyelerini düşürebilir.
Kemik iliği bozuklukları: Kemik iliğinin yeterli nötrofil üretme yeteneğini etkileyen durumlar nötropeniye yol açabilir. Örnekler arasında aplastik anemi, miyelodisplastik sendrom (MDS) ve lösemi yer alır. 4. Otoimmün hastalık: Sistemik lupus eritematozus (SLE) ve romatoid artrit gibi bazı otoimmün hastalıklar, bağışıklık sistemi nötrofillere saldırıp onları yok ettiği için nötropeniye neden olabilir.
Beslenme eksiklikleri: Başta B12 vitamini, folat ve bakır olmak üzere bazı besin maddelerindeki eksiklikler nötrofil üretiminin azalmasına yol açabilir. Nötropeni semptomları, ciddiyetine ve enfeksiyon varlığına bağlı olarak değişebilir. Hafif vakalarda, bireylerde fark edilebilir bir belirti olmayabilir. Bununla birlikte, daha ciddi vakalarda, sık veya ciddi enfeksiyonlara daha yatkın olabilirler. Nötropenisi olan kişilerde yaygın enfeksiyon belirtileri ateş, titreme, boğaz ağrısı, ağız yaraları ve cilt enfeksiyonlarını içerebilir. Nötropeni tedavisi altta yatan nedene ve ciddiyetine bağlıdır. Hiçbir belirti göstermeyen hafif vakalarda özel bir tedaviye ihtiyaç duyulmayabilir. Bununla birlikte, şiddetli veya semptomatik nötropeni vakalarında, odak noktası enfeksiyonu yönetmek ve temel nedeni tedavi etmektir. Tedavi seçenekleri arasında nötrofil üretimini uyarmak için büyüme faktörlerinin kullanılması, ilaçların değiştirilmesi ve altta yatan koşulların yönetilmesi yer alabilir. Nötropenisi olan kişilere genellikle enfeksiyon riskini azaltmak için iyi el hijyeni uygulamak, grip mevsiminde kalabalık yerlerden kaçınmak ve enfeksiyon belirtileri ortaya çıkarsa derhal tıbbi yardım almak gibi önlemler almaları tavsiye edilir. Nötrofil seviyelerini izlemek ve tedaviyi gerektiği gibi ayarlamak için düzenli kan sayımı takibi şarttır.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
“Sitotoksik”, hücrelere zarar verme veya yok etme potansiyeline sahip madde veya ajanları tanımlamak için kullanılan tıbbi bir terimdir. İki kelimenin birleşiminden meydana gelir: Hücreleri ifade eden “cyto” ve zararlı veya zehirli anlamına gelen “toxic”. Sitotoksik maddeler doğal kaynaklı, sentetik veya belirli tıbbi tedavilerin bir sonucu olarak üretilebilir.
Tıpta sitotoksisite, özellikle kanser tedavisi ve terapötiklerinde belirli hücreleri hedef almak ve ortadan kaldırmak için kullanılan birçok ilaç ve tedavinin temel bir özelliğidir. immünosupresif tedavi. Sitotoksisitenin bazı önemli yönleri şunlardır:
Sitotoksik ilaçlar: Sitotoksik ilaçlar, hızla bölünen hücreleri hedef alarak onları kanser hücrelerine karşı etkili hale getiren ilaçlardır. Bu ilaçlar hücre döngüsüne veya DNA replikasyonuna müdahale ederek kanser hücrelerinin ölmesine neden olur. Ne yazık ki sitotoksik ilaçlar, kemik iliği, saç folikülleri ve sindirim sisteminin iç yüzeyinde bulunanlar gibi hızla bölünen normal sağlıklı hücreleri de etkileyebilir. Bu durum, kemik iliği baskılanması, saç dökülmesi ve gastrointestinal rahatsızlık gibi kanser kemoterapisiyle ilişkili yan etkileri açıklamaktadır.
İmmünosupresif sitotoksik ajanlar: Bağışıklık sistemi düzenlemesi bağlamında sitotoksik ajanlar, otoimmün hastalıklarda görüldüğü gibi bağışıklık sisteminin vücudun kendi dokularına saldırdığı durumlarda bağışıklık tepkisini bastırmak için kullanılır. romatoid artrit, lupus ve multipl skleroz gibi salgınlar. Otoimmün yanıttan sorumlu bağışıklık hücrelerini inhibe ederek, bu sitotoksik ajanlar hastalığın kontrolüne yardımcı olabilir, ancak enfeksiyon riskini de artırabilir.
Bağışıklık sistemi üzerindeki sitotoksik etkiler: Sitotoksisite, sitotoksik T lenfositleri (CTL’ler) ve doğal öldürücü (NK) hücreler gibi belirli bağışıklık hücrelerinin vücuttaki enfekte veya anormal hücreleri tanıma ve ortadan kaldırma yeteneğini de ifade edebilir. Bu bağışıklık hücreleri, hedef hücrelerde apoptozu (programlanmış hücre ölümü) tetiklemek için perforin ve granzim gibi toksik maddeler salgılayarak tehdidi etkili bir şekilde ortadan kaldırır.
Sitotoksisite testi: Sitotoksisite testleri, ilaçların, kimyasalların veya diğer maddelerin hücreler üzerindeki potansiyel toksik etkilerini değerlendirmek için kullanılan laboratuvar testleridir. Bu testler, potansiyel tedavilerin güvenliğini ve etkinliğini değerlendirmek için ilaç araştırma ve geliştirmede gereklidir.
Sitotoksisitenin önlenmesi: Sağlık hizmetleri ortamında sitotoksik önlemler, sitotoksik ilaçların hazırlanması, uygulanması ve verilmesi sırasında sağlık çalışanlarını ve hastaları sitotoksik ilaçlara maruz kalmaktan korumak için alınan önlemlerdir. ve imha edin. Bu önlemler, kişisel koruyucu ekipman (KKE) giymeyi ve kazara maruz kalmayı önlemek için özel kullanım prosedürlerini takip etmeyi içerir.
Sitotoksik ajanlar tıbbi tedavide güçlü araçlar olabilirken, kullanımlarının riskler ve potansiyel yan etkiler de taşıdığı unutulmamalıdır. Bu nedenle, genellikle her bir hasta için potansiyel fayda ve riskleri dikkatlice tartan uzman sağlık profesyonellerinin rehberliğinde reçete edilir ve uygulanır.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Histopatoloji, hücresel ve doku seviyelerindeki değişiklikleri ve anormallikleri incelemek için doku örneklerinin mikroskobik incelemesiyle ilgilenen patoloji dalıdır. Tıpta kullanılan önemli bir tanı aracıdır ve hastalığın doğasını ve ilerlemesini anlamanın yanı sıra tedavi stratejilerinin geliştirilmesinde de önemli bir rol oynar.
“Histopatoloji” kelimesi üç bileşenden türetilmiştir: “histo” doku, “patho” hastalık veya anormallik, “logy” ise çalışma veya bilim anlamına gelir. Dolayısıyla histopatoloji, hastalıklı dokuların incelenmesi olarak anlaşılabilir.
Histopatolojik süreç birkaç adımdan oluşur:
Doku örneği toplama: Histopatolojik analiz yapmak için hastadan biyopsi adı verilen küçük bir doku örneği alınır. Bu biyopsi, şüphelenilen hastalığa ve konumuna bağlı olarak cerrahi eksizyon, ince iğne aspirasyonu veya endoskopik biyopsi gibi çeşitli kaynaklardan elde edilebilir.
Montaj: Toplanan doku örneği korunmalı ve daha fazla bozulmaya uğraması önlenmelidir. Bu, immobilizasyon adı verilen bir işlemle sağlanır. En yaygın kullanılan fiksatif, dokulardaki hücresel yapıları ve proteinleri stabilize eden formalindir.
İşlem: Sabitlendikten sonra, kumaş suyu çıkarmak ve yerine parafin mumu koymak için bir dizi adımdan geçer. Bu işleme doku işleme adı verilir ve mikroskop altında incelenebilecek ince kesitlerin oluşturulmasını sağlar.
Mikrotomi: İşlenen doku parafin bloklara gömülür ve tipik olarak 4 ila 5 mikrometre kalınlığında çok ince dilimler bir mikrotom ile kesilir. Bu ince dilimler daha sonra lamlara monte edilir.
Renklendirme: Hücre yapılarının ve spesifik doku bileşenlerinin görünürlüğünü artırmak için doku kesitleri çeşitli boyalarla boyanmıştır. Hematoksilen ve eozin (H&E) boyama en yaygın kullanılan boyama tekniğidir. Hematoksilen çekirdeği mavi/mor boyarken, eozin sitoplazmayı ve hücre dışı yapıyı pembe boyar.
Mikroskobik inceleme: Boyanan doku kesitleri daha sonra bir patolog tarafından optik mikroskop altında incelenir. Patolog hücresel mimariyi, hücre morfolojisini ve anormal hücrelerin, enflamasyonun, enfeksiyonun veya diğer doku değişikliklerinin varlığını analiz eder. Bu gözlemlere dayanarak patolog bir tanı koyar ve ilgili hekime bir rapor sunar.
Histopatoloji, onkoloji, gastroenteroloji, dermatoloji ve diğerleri dahil olmak üzere çeşitli tıbbi uzmanlık alanlarında kullanılır. Diğer durumların yanı sıra kanserlerin, bulaşıcı hastalıkların, otoimmün bozuklukların, enflamatuar durumların ve doğum kusurlarının teşhis edilmesine yardımcı olur.
Özetle, histopatoloji, modern tıpta hastalığı teşhis etmek ve anlamak, tedavi kararlarına rehberlik etmek ve tedavilerin etkinliğini izlemek için doku örneklerinin mikroskobik düzeyde incelenmesini sağlayan önemli bir araçtır. terapi.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Tıbbi açıdan “remisyon”, hastalığın belirti ve semptomlarının geçici veya kalıcı olarak azalması veya ortadan kalkması anlamına gelir. Genellikle kanser, otoimmün hastalıklar ve bazı kronik hastalıklarla ilişkilendirilir. Remisyon kavramı özellikle kanser ve diğer kronik hastalıkların tedavisinde önemlidir, çünkü tedaviye olumlu bir yanıt verildiğini ve hastalık aktivitesinin azaldığını gösterir.
İşte remisyonla ilgili bazı önemli noktalar:
** Kısmi remisyon: ** Bazı durumlarda, “kısmi remisyon” terimi hastalık semptomlarında veya tümör boyutunda önemli bir azalmayı tanımlamak için kullanılır, ancak hastalık henüz tam olarak çözülmemiştir. Bu durum hala devam eden tedavi ve izleme gerektirebilir.
**Tam remisyon: ** “Tam yanıt” veya “tam remisyon” olarak da bilinen bu terim, tespit edilebilir hiçbir hastalık belirtisi olmadığında kullanılır. Bu, hastanın semptomlarının düzeldiği ve tüm tanısal testlerin (örn. görüntüleme testleri, kan testleri) normal olduğu anlamına gelir. Ancak bu, hastalığın tamamen iyileştiği anlamına gelmez; o sırada tespit edilemeyen mikroskobik hastalık kalıntıları hala olabilir.
** İyileşmeye karşı remisyon: ** Remisyon iyileşme ile karıştırılmamalıdır. İyileşme, hastalığın vücuttan tamamen atılmasını içerirken, remisyon sağlığın iyileştiği ve semptomların hafiflediği bir dönem anlamına gelir. Belirli kanser türleri gibi bazı hastalıklar tedavi edilebilirken, diğerleri remisyona girebilir ve semptomlar daha sonra tekrarlayabilir.
**Remisyonda geçen süre: ** Remisyon süresi önemli ölçüde değişebilir. Geçici olabilir ve haftalar, aylar veya yıllar sürebilir. Bazı durumlarda, remisyon süresiz olarak devam ettirilebilir, bu da çok az semptomla veya hiç semptom olmadan kronik hastalığa yol açabilir.
** Remisyon ve kanser: ** Kanser tedavisinde remisyona ulaşmak ana tedavi hedeflerinden biridir. Farklı kanser türlerinde remisyonu belirlemek için tümörün kaybolması, kanserli lezyonların iyileşmesi ve belirli testlerde negatif sonuçlar gibi farklı kriterler vardır. Remisyona girdikten sonra, kanser hastaları genellikle herhangi bir nüks belirtisi olup olmadığını izlemek için düzenli olarak izlenir.
** Spontane remisyon: **Bazen otoimmün hastalıklar ve kanser de dahil olmak üzere bazı hastalıklar spesifik bir tedavi olmaksızın remisyona girebilir. Buna “spontan remisyon” veya “kendiliğinden remisyon” denir. Bu fenomenin altında yatan mekanizmalar her zaman iyi anlaşılmış değildir.
**Remisyonun başlangıcı: **Remisyon, cerrahi, kemoterapi, radyasyon tedavisi, immünoterapi, hedefe yönelik tedavi ve ilaç tedavisi dahil olmak üzere çeşitli tedavilerle indüklenebilir. Tedavi seçimi spesifik hastalığa ve evresine bağlıdır.
Özetle remisyon, tıbbi bağlamda bir hastalığın semptomlarının veya belirtilerinin değişen derecelerde şiddet ve süreyle hafiflemesini veya ortadan kalkmasını tanımlamak için kullanılan bir terimdir. Kanser de dahil olmak üzere çeşitli kronik hastalıkların yönetiminde olumlu bir sonuçtur ve ilgili kişinin sağlık ve yaşam kalitesinde bir iyileşme dönemine işaret eder. Remisyon dönemlerinde herhangi bir nüksetme ya da nüksetme belirtisi olup olmadığını izlemek için düzenli tıbbi takip şarttır.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Tümör belirteçleri, hem normal hem de kanser hücreleri tarafından üretilen ve belirli kanserleri veya diğer hastalıkları olan kişilerin kanında, idrarında veya dokusunda tespit edilebilen maddelerdir. Bu belirteçler kanser hastalarının tespiti, tedavisi ve izlenmesinde teşhis ve prognostik araçlar olarak kullanılmaktadır. Bununla birlikte, tümör belirteçlerinin tek başına kesin tanı araçları olmadığını ve kullanımlarının genellikle diğer laboratuvar testleri ve klinik değerlendirmelerle birleştirildiğini anlamak önemlidir.
İşte tümör belirteçleri hakkında bazı önemli noktalar:
Üretim ve radyasyon: Tümör belirteçleri genellikle kanser hücrelerinin kendileri tarafından üretilen veya kanser tümörleri tarafından kan dolaşımına veya vücut sıvılarına salınan proteinler veya diğer maddelerdir. Bazen bu belirteçler, vücudun kanserin varlığına verdiği yanıtın bir parçası olarak bazı normal hücreler tarafından da üretilebilir.
Tanı ve prognozda kullanım: Tümör belirteçleri esas olarak onkolojide kanserin teşhisine, evrelendirilmesine ve ilerlemesinin veya tedavinin etkinliğinin izlenmesine yardımcı olmak için kullanılır. İyi huylu (kanserli olmayan) ve kötü huylu (kanserli) hastalık arasında ayrım yapılmasına yardımcı olabilir ve hastalığın kapsamı ve invazivliği hakkında bilgi sağlayabilirler.
Tedavi sonrası takip: Tedavi gören kanser hastaları için tümör belirteçleri tedaviye yanıtı değerlendirmek için kullanılabilir. Zaman içinde belirteç seviyelerindeki değişiklikler tedavinin işe yarayıp yaramadığını veya kanserin ilerleyip ilerlemediğini gösterebilir.
Sınır: Tümör belirteçleri değerli araçlar olmalarına rağmen sınırlamaları da vardır. Tüm kanserler tespit edilebilir seviyelerde tümör belirteçleri üretmez ve bazen kanser dışı durumlar belirteç seviyelerini yükselterek pozitif bir sonuca yol açabilir. Yanlışlık. Bu nedenle, tümör belirteçlerinin kullanımı her kanser türüne özgüdür ve bunların yorumlanması klinik bulgular ve diğer tanı testleriyle birlikte yapılmalıdır.
Yaygın kanser belirteçleri: Yaygın olarak kullanılan tümör belirteçlerine bazı örnekler aşağıda verilmiştir:
Prostat Spesifik Antijen (PSA): Prostat kanserini tespit etmek ve izlemek için kullanılır.
Karsinoembriyonik antijen (CEA): Bazı gastrointestinal ve diğer kanserlerde görülür.
Alfa-fetoprotein (AFP): Bazı karaciğer ve germ hücreli tümörlerde yükselir. -CA-125:
Yumurtalık kanseri ile ilişkilidir. AC 19-9: Pankreas ve gastrointestinal kanserler ile ilişkilidir.
Bireysel varyasyon: Tümör belirteci seviyelerinin kişiden kişiye büyük farklılıklar gösterebileceğini ve her zaman doğrudan tümör boyutu veya evresi ile ilişkili olmadığını unutmamak önemlidir. Bazı kanser hastaları yüksek belirteç seviyelerine sahip olmayabilirken, hafif hastalığı olan diğerlerinde anormal seviyeler olabilir.
Özetle, tümör belirteçleri kanser teşhisi ve tedavisinde önemli bir rol oynamaktadır. Bununla birlikte, hastanın durumunu doğru bir şekilde değerlendirmek için kullanımları her zaman diğer klinik ve tanısal testlerle birleştirilmelidir. Tümör belirteç sonuçlarının yorumlanması onkoloji alanında kalifiye tıp uzmanları tarafından yapılmalıdır.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
İnflamazom, bağışıklık sisteminin enfeksiyon ve doku hasarına verdiği yanıtta önemli bir rol oynadığı bilinen çok proteinli kompleksidir. Doğuştan gelen bağışıklık sisteminin bir parçası olarak, patojenlere karşı vücudun ilk basamak savunma mekanizmasını oluştururlar.
Vücut yabancı istilacıları veya doku hasar gördüğünde salınan hasarla ilişkili molekülleri (DAMP’ler) algıladığında, inflamazomlar aktive olur. IL-1β (Interleukin-1 beta) ve IL-18 (Interleukin-18) dahil olmak üzere proinflamatuar sitokinlerin üretimine yol açan bir sinyal yolları zincirini indüklerler.)
Çekirdek bileşenleri sensör proteinleri, adaptör proteinleri ve efektör proteinlerini içerir. Tanımlanmış çeşitli inflamazom türleri vardır, NLRP3 inflamazomu (NACHT, LRR ve PYD alanlarını içeren protein 3) en çok çalışılan ve yaygın olarak tanınanlardan biridir.
NLRP3 inflamazom aktivasyonu, patojenle ilişkili moleküler kalıplar (PAMP’ler), DAMP’ler ve stresle ilişkili diğer moleküller dahil olmak üzere geniş bir sinyal yelpazesi tarafından indüklenir. NLRP3 yazımı aktive olduktan sonra, pro-IL-1β ve pro-IL-18’in aktif formlarına bölünmesinde işlev gören ve olgun sitokinlerin salınmasıyla sonuçlanan inflamatuar kaspaz olan kaspaz-1’i çeker ve aktive eder.
İnflamazomlar “kanonik” ve “kanonik olmayan” olarak ikiye ayrılır. “Kanonik” kaspaz-1 bağımlı süreci ifade ederken, inflamazomların “kanonik olmayan” aktivasyonu ya kaspaz-11 (farelerde) ya da kaspaz-4/5 (insanlarda) yolu olarak tanımlanır.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Adams-Oliver sendromu doğumda ortaya çıkan nadir bir durumdur. Başlıca özellikleri derinin disgenezisi (konjenital kutanöz hipoplazi olarak adlandırılır) ve uzuvların malformasyonlarıdır. Adams-Oliver sendromlu kişilerde çeşitli başka semptomlar da görülebilir.
Adams-Oliver sendromlu kişilerin çoğunda konjenital deri hipoplazisi vardır. Bu, genellikle başın üst kısmında (kafatası) görülen fokal cilt kaybı alanları ile karakterize bir durumdur. Bazı durumlarda, deri altındaki kemik de az gelişmiştir. Bu rahatsızlığa sahip kişilerde genellikle yara izi ve etkilenen bölgede saç büyümesi eksikliği görülür.
Adams-Oliver sendromlu kişilerde el ve ayak anormallikleri de yaygındır. Bunlar en sık el ve ayak parmaklarını etkiler ve anormal tırnakları, birlikte büyüyen el veya ayak parmaklarını (sindaktili) ve anormal derecede kısa veya eksik el veya ayak parmaklarını (brakidaktili veya oligodaktili) içerebilir. Bazı durumlarda ellerdeki, ayaklardaki ve alt bacaklardaki diğer kemikler de deforme olabilir veya eksik olabilir.
Etkilenen bazı bebekler konjenital kutanöz telenjiektazi adı verilen bir durumdan muzdariptir. Kan damarlarındaki bu tahribat, cildin kırmızımsı veya morumsu, ağ benzeri bir desen geliştirmesine neden olur. Ayrıca, Adams-Oliver sendromlu kişilerde kalp ve akciğerler arasındaki kan damarlarında (pulmoner hipertansiyon) yaşamı tehdit edebilecek yüksek kan basıncı görülebilir. Etkilenen bireylerde başka vasküler sorunlar ve kalp kusurları gelişebilir.
Adams-Oliver sendromlu kişilerde gelişimsel gecikmeler, öğrenme güçlükleri ve beyin yapısında anormallikler gibi nörolojik sorunlar görülebilir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Krippel-Feil Sendromu, iki veya daha fazla servikal omurun doğuştan kaynaştığı nadir bir durumdur. Krippel-Feil Sendromu olan kişiler boyunda sınırlı hareketlilik ve ağrı yaşayabilir.
Klippel-Feil sendromunun en yaygın belirtileri şunlardır:
Kısa boyun, muhtemelen düşük oksipital saç çizgisi
Yüz, boyun, gövde ve sırtı etkileyen sınırlı esneklik ve hareketlilik
Ağrı – Kaynaşmış omurlar sinir hasarına ve baş, boyun ve sırtta ağrıya neden olabilir.
İşitme kaybı – ses sinyalleri kulağa veya beynin doğru kısmına ulaşmakta zorluk çeker
Belirli gen mutasyonlarına sahip kişilerde bu durumun gelişme riski yüksektir. GDF6 (büyüme farklılaşma faktörü 6) veya GDF3 (büyüme farklılaşma faktörü 3) genlerindeki mutasyonlar bu bozukluğa neden olabilir.
Doktorlar Klippel-Feil sendromunu genellikle doğumda veya doğumdan hemen önce gözlem yoluyla teşhis eder. Bozukluğun hafif veya şiddetli olup olmadığını öğrenmek için testler yapılabilir.
Doktorlar artık Klippel-Feil sendromunu boyundaki kemikleri düzeltmek için ameliyatla tedavi ediyor. Diğer semptomları tedavi etmek için çeşitli doktorlara görünebilirsiniz.
Krippel-Feil Sendromu olan kişiler boyuna zarar verebilecek aktivitelerden kaçınmalıdır. Uygun tedavi ve bakım ile Klippel-Feil sendromu olan kişiler normal yaşamlarını sürdürebilirler. Görünüm, vakanın ciddiyetine ve kişinin sahip olduğu ilişkili tıbbi durumların sayısına bağlıdır.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Kemik Mahzumları (Osteofitler, kemiklerin uçları boyunca gelişen kemik çıkıntılarıdır. Osteofitler (osteofitler) genellikle kemiklerin bir eklem içinde birleştiği yerde oluşur. Omurga kemiklerinde de oluşabilirler.
Osteofitlerin ana nedeni osteoartrit ile ilişkili eklem hasarıdır. Osteofitlerin çoğu hiçbir belirtiye neden olmaz ve yıllarca fark edilmeyebilir. Tedavi gerekli olmayabilir. Tedaviye ihtiyacınız olup olmadığı sporların nerede bulunduğuna ve sağlığınızı nasıl etkilediğine bağlıdır.
Semptomlar
Osteofitlerin çoğu hiçbir belirti veya semptoma neden olmaz. Başka bir hastalıktan dolayı çekilen röntgende bir büyüme ortaya çıkana kadar kemik çıkıntınız olduğunu fark etmeyebilirsiniz. Ancak bazı durumlarda osteofitler eklem ağrısına ve hareket kısıtlılığına neden olabilir. Spesifik semptomlar osteofitlerin bulunduğu yere bağlıdır.
Örnekler şunları içerir:
Dizler: Dizdeki kemik çıkıntıları bacak uzatıldığında veya büküldüğünde ağrıya neden olabilir.
Omurga: Omurgada, osteofitler omuriliği içeren alanı daraltabilir. Bu kemik çıkıntıları omuriliği veya sinir köklerini sıkıştırarak kollarda ve bacaklarda güçsüzlük ve uyuşukluğa neden olabilir.
Kalça: Kemik çıkıntıları kalça hareketlerinde ağrıya neden olabilir, ancak dizde de ağrıya neden olabilirler. Pozisyona bağlı olarak, osteofitler kalça ekleminin hareket aralığını sınırlayabilir.
Ne zaman bir doktora görünmeliyim?
Bir veya daha fazla eklemde ağrı veya şişlik varsa ya da bir eklemi hareket ettirmekte zorlanıyorsanız doktorunuza görünün. neden
Osteoartrite bağlı eklem hasarı osteofitlerin en yaygın nedenidir. Osteoartrit kemiklerin uçlarını yastıklayan kıkırdağı tahrip ettiğinde, vücut hasarlı bölgelerin yakınında osteofit oluşturarak kaybı onarmaya çalışır.
Risk faktörü
Artriti olan kişilerde kemik çıkıntısı riski artar.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Mitokondri, insan hücreleri de dahil olmak üzere çoğu ökaryotik hücrede bulunan özelleşmiş organellerdir. Birincil işlevleri hücresel solunum adı verilen bir süreçle adenozin trifosfat (ATP) formunda enerji üretmek olduğu için bazen hücrenin “enerji santralleri” olarak adlandırılırlar.
Mitokondri
Mitokondri, nerede ve işlevi nedir
Mitokondrilerin mitokondriyal DNA (mtDNA) adı verilen kendi DNA’ları vardır ve hücre içinde bağımsız olarak çoğalabilirler. Milyarlarca yıl önce ökaryotik progenitör hücreler tarafından yutulan eski simbiyotik bakterilerden kaynaklandıkları düşünülmektedir.
Mitokondrinin ana işlevi, sitrik asit döngüsü (Krebs döngüsü veya TCA döngüsü olarak da adlandırılır) ve oksidatif fosforilasyon olarak bilinen bir dizi biyokimyasal reaksiyon yoluyla ATP üretmektir. Bu süreçler, enzimlerin ve elektron taşıma zincirlerinin bulunduğu mitokondriyal iç zar içinde gerçekleşir.
Enerji üretimine ek olarak, mitokondri diğer bazı hücresel süreçlerde de rol oynar. Kalsiyum sinyali, hücre büyümesi ve farklılaşması, hücre ölümünün düzenlenmesi (apoptoz) ve hücre fonksiyonu için gerekli spesifik moleküllerin sentezinde rol oynarlar.
Mitokondriyal işlev bozukluğu veya hasar, mitokondriyal bozukluklar, nörodejeneratif bozukluklar, metabolik bozukluklar ve yaşa bağlı hastalıklar dahil olmak üzere çeşitli hastalıklara ve durumlara yol açabilir. Bilim insanları sağlık ve hastalıktaki rollerini keşfetmeye devam ettikçe mitokondri ve işlevleri üzerine araştırmalar devam etmektedir.
Mitokondriyal DNA (mtDNA) kalıtımı
Mitokondriyal DNA (mtDNA) kalıtımı, mtDNA’nın bir nesilden diğerine aktarıldığı süreci ifade eder. Her iki ebeveynden de kalıtılan nükleer DNA’nın aksine, mtDNA yalnızca anneden kalıtılır. Bu kalıtım modeli maternal veya anneden kalıtım olarak bilinir.
Bu benzersiz kalıtım modelinin nedeni, spermin döllenme sırasında genellikle döllenmiş yumurtaya mitokondri bağışlamamasıdır. Bunun yerine, döllenmiş yumurta içinde bulunan mitokondriler yalnızca annenin yumurtasından türetilir. Bu nedenle, yavrunun tüm mtDNA’sı anneden gelir.
Döllenme sırasında, spermde mitokondri varsa, bunlar normalde ubikitinasyon adı verilen bir işlemle yok edilir. Bu, ortaya çıkan embriyoların yalnızca maternal mtDNA taşımasını sağlar. Sonuç olarak, ara sıra mutasyonlar meydana gelse de mtDNA dizileri nesiller boyunca nispeten sabit kalır.
mtDNA sadece anneden kalıtıldığı için, doğrudan anne soyunu takip eder. Bu özellik mtDNA’yı insan evrimi, popülasyon genetiği çalışmaları ve anne soyunun takibi için kullanışlı hale getirir. Bilim insanları, bir bireyin mtDNA dizilerini karşılaştırarak ortak anne soyunun izini sürebilir ve ortak anne soyunu paylaşan insan grupları olan mitokondriyal haplogruplarını oluşturabilir.
Her ne kadar mtDNA öncelikle anneden miras alınsa da, mtDNA’nın babadan miras alındığı nadir vakaların rapor edildiğini belirtmek önemlidir. Bununla birlikte, bu vakalar çok nadirdir ve genellikle
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Periferik vasküler hastalık olarak da adlandırılan periferik arter hastalığı, kalp dışındaki arterlerin, özellikle bacaklarda ve ayaklarda daralması veya tıkanması ile karakterize bir durumdur. Genellikle aterosklerozdan, arter duvarlarında yağ birikintilerinin (plak) oluşmasından kaynaklanır ve ekstremitelere kan akışının azalmasına yol açar.
PAD gelişimi, arter duvarlarında kronik inflamasyon ve plak birikimi ile ilişkilidir. Sigara, yüksek tansiyon, yüksek kolesterol ve diyabet gibi risk faktörleri arterlerin iç yüzeyinin hasar görmesine katkıda bulunur. Bu hasar enflamatuar bir yanıtı tetikleyerek kolesterol, kalsiyum ve diğer maddelerin arter duvarlarında birikmesine neden olur. Zamanla bu plaklar arterleri daraltarak kan akışını kısıtlar ve bacak kaslarına ve dokularına oksijen ve besin tedarikini azaltır.
Belirtiler ve bulgular
PAD’nin en yaygın semptomu, fiziksel aktivite sırasında bacak ağrısı, kramplar ve yorgunluk anlamına gelen aralıklı topallamadır. Bu durum genellikle baldır kaslarını etkiler, ancak uyluk ve kalçaları da etkileyebilir. Ağrı dinlenmeyle azalır ve yürümeye başladığınızda geri döner. Diğer belirti ve semptomlar arasında uyuşukluk, halsizlik, soğuk bacaklar, cilt değişiklikleri (parlak veya renksiz cilt gibi), ayaklarda ve ayak parmaklarında yavaş iyileşen yaralar veya ülserler, zayıf ayak tırnağı büyümesi ve ayaklarda nabız sayılabilir. Zayıf veya hiç nabız yok, ereksiyon. Azalmaya bağlı işlev bozukluğu
PAD tedavisi semptomları hafifletmeyi, kan akışını iyileştirmeyi ve komplikasyonları önlemeyi amaçlar. Yaklaşımlar şunları içerir:
Yaşam tarzı değişiklikleri: Sigarayı bırakmak, sağlıklı beslenmek, düzenli egzersiz yapmak ve yüksek tansiyon, yüksek kolesterol ve diyabet gibi altta yatan tıbbi durumları yönetmek.
Dozajlama: Reçete edilen ilaçlar arasında kan pıhtılaşması riskini azaltmak için antiplatelet ilaçlar, kolesterolü düşürmek için statinler, tansiyon ilaçları ve yürüme mesafesini artırmak için ilaçlar yer alabilir. 3. Anjiyoplasti ve stentleme: Anjiyoplasti, balon uçlu bir kateterin etkilenen artere yerleştirildiği ve daralmış alanı genişletmek ve kan akışını iyileştirmek için şişirildiği bir prosedürdür. Bazı durumlarda, arteri açık tutmak için bir stent (küçük bir ağ tüpü) kullanılır.
Bypass ameliyatı: Anjiyoplastinin uygulanabilir veya etkili olmadığı ciddi vakalarda bypass ameliyatı önerilebilir. Bu, kan akışını yeniden sağlamak için arterin tıkalı kısmını bypass eden sağlıklı kan damarlarından veya sentetik hortumdan bir greft oluşturmayı içerir.
Ayırıcı tanı
PAD benzeri semptomlarla ortaya çıkabilen bozukluklar arasında nöropatik klodikasyon (spinal stenoza bağlı), kas-iskelet sistemi bozuklukları (artrit ve sinir sıkışması gibi), venöz yetmezlik, periferik nöropati ve derin ven trombozu bulunmaktadır. PAD’yi bu diğer durumlardan ayırt etmek için bir tıp uzmanı tarafından doğru tanı konulması şarttır.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Histeroskopi nedir? Histeroskopi, rahmin (uterus) içine bakmak için histeroskop adı verilen ince, ışıklı bir tüpün vajina ve servikse yerleştirildiği tıbbi bir prosedürdür. Histeroskop, doktorların rahim ve fallop tüplerinin açıklıklarını görmesini sağlar.
İki tür histeroskopi vardır: Tanısal histeroskopi ve cerrahi histeroskopi.
Histeroskopi ile tanı: Bu prosedür, belirli bir jinekolojik durumun veya hastalığın nedenini araştırmak için yapılır. Bu, doktorların anormal uterin kanama, infertilite, tekrarlayan düşükler veya uterusta fibroid, polip veya adezyon (yara dokusu) varlığı gibi sorunları tanımlamasına yardımcı olur. Histeroskopi teşhisi sırasında doktorunuz daha ileri inceleme için doku örnekleri de alabilir (biyopsi).
Histeroskopik cerrahi: Görselleştirmeye ek olarak, cerrahi histeroskopi doktorların rahim içinde cerrahi prosedürler gerçekleştirmesine olanak tanır. Bu, poliplerin, fibroidlerin veya yapışıklıkların giderilmesini ve uterin septumun (uterusun duvarlarla bölündüğü konjenital bir bozukluk) tedavisini içerebilir. Bu prosedürleri gerçekleştirmek için doktorlar histeroskop aracılığıyla yerleştirilen özel aletler kullanırlar.
Histeroskopi genellikle ameliyatın karmaşıklığına bağlı olarak genel veya lokal anestezi kullanılarak ayakta tedavi bazında yapılır. İyileşme süresi nispeten kısadır ve çoğu kadın 1-2 gün içinde normal faaliyetlerine dönebilir. Bununla birlikte, işlemden birkaç gün sonra hafif kramp ve hafif vajinal kanama meydana gelebilir.
Her tıbbi prosedür gibi histeroskopinin de potansiyel riskleri ve komplikasyonları vardır, ancak bunlar genellikle nadirdir. Bunlar arasında enfeksiyon, kanama, rahim veya rahim ağzında hasar ve anesteziden kaynaklanan komplikasyonlar yer alabilir. Doktorunuz bu riskleri sizinle tartışacak ve ameliyattan önce sahip olabileceğiniz endişeleri ele alacaktır.
Histeroskopinin özel tıbbi durumunuz için uygun olup olmadığını belirlemek ve potansiyel faydaları ve riskleri ayrıntılı olarak tartışmak için kalifiye bir doktora danışmanız önemlidir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Ortoloji, en son ortak atanın ortak atasal geninden türetilen farklı türlerdeki genler arasındaki ilişkileri ifade eder. İki genin ortolog olduğu düşünüldüğünde, farklı türlerde benzer işlevleri ve evrimsel ilişkileri korudukları anlamına gelir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Artralji, vücudun bir veya daha fazla ekleminde rahatsızlık, ağrı veya sızı olarak tanımlanır. Hafiften şiddetliye kadar değişebilir ve akut (kısa süreli) veya kronik (uzun süreli) olabilir. Eklem ağrısına yaralanma, iltihaplanma, enfeksiyon ve altta yatan hastalık gibi birçok faktör neden olabilir. Eklem ağrısının tanımı, belirtileri ve bulguları, teşhisi, tedavisi ve ayırıcı tanısı hakkında daha fazla bilgiyi burada bulabilirsiniz.
Tanım:
Artralji, eklemlerdeki rahatsızlık, ağrı veya acıdır. Eklemler, harekete izin veren ve destek sağlayan kemikler arasındaki bağlantılardır. Eklem ağrısı dizler, kalçalar, omuzlar, bilekler ve ayak bilekleri dahil olmak üzere vücuttaki herhangi bir eklemi etkileyebilir.
Belirtiler ve İşaretler:
Eklem ağrısı ile ilişkili semptom ve bulgular altta yatan nedene bağlı olarak değişir, ancak genellikle şunlardır:
Ağrı: Bir veya daha fazla eklemde ağrı, zonklayıcı ağrı. veya bıçak saplanır gibi bir ağrı.
Şişme: Eklem iltihabı, etkilenen eklem çevresinde şişlik, kızarıklık veya sıcaklığa neden olabilir.
Sertlik: Özellikle hareketsizlik veya dinlenme dönemlerinden sonra sınırlı hareket aralığı.
Zayıflık: Eklem hareketlerini gerçekleştirmede veya ağırlık taşımada zorluk veya güçsüzlük.
Kilitlenme veya Tıklama Hissi: Bazı kişiler hareket sırasında eklemlerinde kilitlenme veya tıklama hissi yaşayabilir.
Sınırlı hareketlilik: Eklem ağrısı esnekliği sınırlayabilir ve günlük aktiviteleri gerçekleştirmeyi zorlaştırabilir.
Teşhis:
Eklem ağrısının nedenini teşhis etmek Eklem ağrısı bir doktor tarafından kapsamlı bir muayene gerektirir.
Teşhis süreci şunları içerir:
Tıbbi Geçmiş: Sağlık uzmanınız eklem ağrınızın başlangıcı, süresi ve özelliklerinin yanı sıra ilişkili semptomlar ve önceki yaralanmalar hakkında sorular soracaktır.
Fiziksel Muayene: Etkilenen eklemler şişlik, hassasiyet, hareketlilik ve iltihap belirtileri açısından incelenecektir.
Görüntüleme: Eklemi ve çevresindeki yapıları görüntülemek ve herhangi bir anormalliği tespit etmek için röntgen, MRI veya BT taraması istenebilir.
Laboratuvar Testleri: Kan testleri iltihap, enfeksiyon veya romatoid artrit gibi belirli otoimmün hastalıkların belirteçlerini tanımlamaya yardımcı olabilir.
Tedavi:
Eklem ağrısının tedavisi altta yatan nedene bağlıdır. Yaygın tedavi yaklaşımları şunları içerir:
İlaç tedavisi: Reçetesiz satılan ağrı kesiciler (asetaminofen, non-steroid anti-inflamatuar ilaçlar gibi) hafif ve orta dereceli eklem ağrısının yönetilmesine yardımcı olabilir. Bazı durumlarda, kortikosteroidler, hastalığı modifiye edici anti-enflamatuar ilaçlar (DMARD’lar) ve enflamatuar bozukluklar için biyolojikler gibi reçeteli ilaçlar gerekebilir.
Fizik Tedavi: Bir fizyoterapist tarafından reçete edilen egzersizler ve terapiler eklem hareketliliğini artırmaya, destekleyici kasları güçlendirmeye ve ağrıyı azaltmaya yardımcı olabilir.
Yaşam Tarzı Değişiklikleri: Kilo yönetimi, düzenli egzersiz ve uygun vücut mekaniği eklem stresini azaltabilir ve genel eklem sağlığını iyileştirebilir.
Yardımlar: Etkilenen eklemi desteklemek ve hareketliliği artırmak için ateller, diş telleri veya yürüme yardımcıları önerilebilir.
Enjeksiyonlar: Enflamasyonu ve ağrıyı azaltmak için doğrudan eklem içine kortikosteroid enjeksiyonları veya hyaluronik asit enjeksiyonları yapılabilir.
Ameliyat: Ağır vakalarda veya konservatif önlemler başarısız olduğunda, eklem replasmanı veya artroskopi gibi cerrahi müdahaleler düşünülebilir.
Ayırıcı Tanı:
Ayırıcı tanıda eklem ağrısına neden olabilecek çeşitli rahatsızlıklar göz önünde bulundurulur. Eklem ağrısı için yaygın ayırıcı tanılar şunlardır:
Osteoartrit: Eklem kıkırdağının tahrip olması ve bunun sonucunda ağrı, sertlik ve şişlik ile karakterize dejeneratif bir eklem hastalığı.
Romatoid artrit: otoimmün bir hastalıktır
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Primer adrenal yetmezlik veya hipoadrenal yetmezlik olarak da bilinen Addison hastalığı, adrenal bezler tarafından yetersiz hormon üretimi ile karakterize nadir bir durumdur. Böbreklerin üst kısmında bulunan bu bezler, çeşitli vücut fonksiyonlarının düzenlenmesinde gerekli olan kortizol ve aldosteron gibi önemli hormonları üretir.
Burada Addison hastalığının tanımı, belirti ve bulguları, teşhisi, tedavisi ve ayırıcı tanısına genel bir bakış yer almaktadır.
Tanımlar:
Addison hastalığı, böbrek üstü bezleri hasar gördüğünde ve artık yeterli kortizol ve aldosteron üretemediğinde ortaya çıkar. En yaygın nedeni, vücudun bağışıklık sisteminin yanlışlıkla adrenal bezlere saldırdığı ve zarar verdiği bir otoimmün reaksiyondur. Diğer nedenler arasında enfeksiyonlar, kanser, bazı ilaçlar ve genetik faktörler yer alır.
Belirtiler ve Bulgular:
Addison hastalığının belirtileri belirsizdir ve yavaş yavaş gelişir. Bunlar genellikle şunları içerir:
Addison hastalığının teşhisi tıbbi öykü, fizik muayene ve laboratuvar testlerinin bir kombinasyonunu gerektirir. Aşağıdaki tanısal adımlar yaygın olarak kullanılır:
Kan testi: Kortizol ve adrenokortikotropik hormon (ACTH) seviyelerini ölçer.
Görüntü incelemesi: Adrenal bezlerin yapısını değerlendirmek için adrenal bezlerinin BT veya MRI taraması yapılabilir.
Tedavi:
Addison hastalığı için tedavinin temel dayanağı, eksik hormonların yerini alan hormon replasman tedavisidir. Bu genellikle kortizol yerine hidrokortizon, prednizon ve deksametazon gibi oral kortikosteroidlerin alınmasını içerir. Aldosteronu yerine koymak için fludrokortizon gibi mineralokortikosteroidlerle ikame de gerekli olabilir. Dozaj bireysel ihtiyaçlara göre ayarlanır ve düzenli izleme gerektirir. Ek olarak, stres yönetimi ve tuz alımının artırılması gibi diyet değişiklikleri önerilebilir.
Ayırıcı tanı:
Ayırıcı tanı önemlidir çünkü birçok hastalık Addison hastalığına benzer semptomları paylaşabilir.
Ayırıcı tanılar şunları içerir:
Sekonder adrenal yetmezlik: Hipofiz veya hipotalamik fonksiyon bozukluğu kortizol gibi hormon üretimini etkiler.
Kronik Yorgunluk: Dinlenme ile düzelmeyen ve genellikle diğer semptomların eşlik ettiği şiddetli yorgunluk ile karakterizedir.
Depresyon: Depresyonda ruh hali değişimleri ve yorgunluk görülür, ancak hiperpigmentasyon ve elektrolit dengesizlikleri mevcut değildir.
Böbreküstü bezlerinin diğer hastalıkları: Adrenal tümörler, adrenal kanama ve konjenital adrenal hiperplazi adrenal yetmezliğe yol açabilir. Addison hastalığınız olduğundan şüpheleniyorsanız veya endişe verici belirtileriniz varsa, doğru teşhis ve uygun tedavi için doktorunuza görünmeniz önemlidir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Hutchinson sendromu olarak da bilinen Hutchinson triadı, Treponema pallidum bakterisinin neden olduğu cinsel yolla bulaşan bir hastalık olan konjenital sifiliz ile ilişkili bir dizi semptomu ifade eder. Adını, 19. yüzyılın sonlarında üçlüyü ilk kez tanımlayan İngiliz cerrah Jonathan Lord Hutchinson’dan almıştır.
Üç özellikten oluşmaktadır.
Tırtıklı veya çentikli dişler: Hutchinson dişleri, rahimde ya da çocukluk döneminde frengi hastalığına yakalanan kişilerde ortaya çıkan diş anormallikleridir. Dişler genellikle normalden daha küçük ve daha geniş aralıklıdır, tırtıklı bir oklüzal yüzeye ve belirgin bir “sürücü şekilli” görünüme sahiptir.
İnterstisyel keratit: Gözün ön tarafındaki saydam kısım olan korneanın iltihaplanması anlamına gelir. Stromal keratit bulanık görme, ışığa karşı hassasiyet ve göz ağrısı gibi görme sorunlarına neden olabilir. Bu, sifiliz enfeksiyonunun korneaya verdiği bağışıklık aracılı hasarın bir sonucudur.
Sekizinci sinir felci: Vestibüler koklear sinir olarak da bilinen sekizinci kraniyal siniri etkileyen bir tür sensörinöral işitme kaybıdır. Bir veya iki kulakta kısmi veya tam işitme kaybına neden olabilir. Hutchinson triadı ile ilişkili işitme kaybı genellikle geri döndürülemez. Hutchinson üçlüsünün doğuştan gelen frengiye özgü olduğunu ve yetişkinlikte ortaya çıkan edinilmiş frengiye özgü olmadığını belirtmek önemlidir. Konjenital sifiliz tedavi edilmediğinde, üçlünün ötesinde kemik anormallikleri, sinir hasarı ve organ hasarı da dahil olmak üzere birçok ciddi komplikasyona yol açabilir.
Frengi önlenebilir ve tedavi edilebilir bir enfeksiyon olduğundan, konjenital frengi ve Hutchinson üçlüsünün gelişmesini önlemek için hamilelik sırasında düzenli tarama ve zamanında tedavi şarttır.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Hertoge belirtisi veya Kraliçe Anne belirtisi olarak da bilinen Hertoge belirtisi, şiddetli hipotiroidizm (tiroid bezinin az çalışması) ile ilişkili fiziksel bir bulgudur. Adını Belçikalı endokrinolog Dr. Eugène Hertoge’dan alan işaret ilk olarak Eugène Hertoge tarafından tanımlanmıştır.
Hertoge belirtisi, kaşların dış üçte birlik kısmının incelmesi veya kaybolması anlamına gelir. Bu işaret kaşların, özellikle de yan (dış) uçlarının seyrekleşmesine ve karakteristik “üçgen” bir görünüm kazanmasına neden olur. Kaşlardaki bu kıl kaybı veya incelme simetrik veya asimetrik olabilir ve genellikle kadınlarda daha belirgindir. Hertogen belirtisinin şiddetli hipotiroidizme işaret edebileceğini ancak kesin bir tanı göstergesi olmadığını unutmamak önemlidir. Hipotiroidi tanısını doğrulamak için diğer belirti ve semptomlar ile tiroid hormonu seviyelerini ölçmek için kan testleri gereklidir.
Tiroidinizin az çalıştığından şüpheleniyorsanız veya kaş değişiklikleri ya da diğer belirtiler konusunda endişeleriniz varsa doğru değerlendirme, tanı ve tedavi için bir sağlık uzmanına başvurmanız önerilir. B. Birinci basamak doktorunuz veya endokrinoloğunuz.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Tahriş edici maddeye tekrar maruz kalınırsa tekrarlama
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
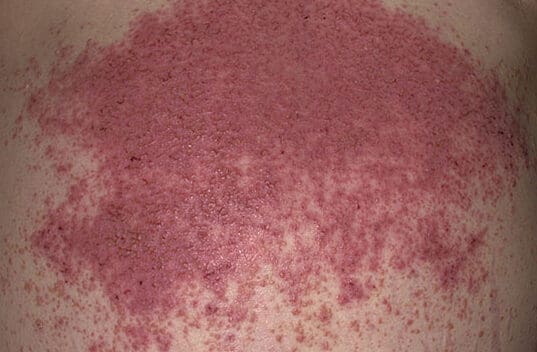
Dermatitis herpetiformis (DH) şiddetli kaşıntı ve kabarcıklı deri lezyonları ile karakterize kronik otoimmün bir deri hastalığıdır. Gluten tüketimine karşı otoimmün bir yanıt olan çölyak hastalığının kutanöz bir belirtisi olduğuna inanılmaktadır.
Madhero88, CC BY-SA 3.0 https://creativecommons.org/licenses/by-sa/3.0, via Wikimedia Commons
Patofizyoloji
Dermatitis herpetiformisin kesin nedeni tam olarak anlaşılamamıştır, ancak otoimmün bir reaksiyon içerdiği bilinmektedir. Genetik olarak yatkın bir kişi glüten tükettiğinde, bağışıklık sistemi glüten proteinlerine karşı antikorlar, özellikle de immünoglobulin A (IgA) antikorları üretir. Bu antikorlar yanlışlıkla ciltteki küçük kan damarlarına saldırarak karakteristik cilt lezyonlarına ve iltihaplanmaya neden olur:
Dermatitis herpetiformis tanısı genellikle laboratuvar testleri, deri biyopsileri ve serolojik testlerin bir kombinasyonunu içerir. Teşhis süreci aşağıdaki adımları içerebilir:
Laboratuvar testleri
Dermatologlar deri lezyonlarını görünümlerini, dağılımlarını ve ilişkili semptomları göz önünde bulundurarak inceler.
Deri biyopsisi
Lezyona bitişik etkilenmemiş deriden küçük bir deri biyopsisi alınır. Biyopsi, cilt papillalarında IgA birikintileri olup olmadığını görmek için mikroskop altında incelenecektir.
Serolojik test
Kan testleri, aşağıdakiler gibi belirli antikorların seviyelerini ölçmek için yapılır: B. Anti-doku transglutaminaz (anti-tTG) ve anti-endomizyal antikor (EMA). Bu antikorların yüksek seviyeleri altta yatan bir gluten intoleransı veya çölyak hastalığına işaret edebilir.
Gluten Testi:
Bazı durumlarda, bir gluten testi bile yapabilirsiniz. Bu, glutenin bir süre için diyete yeniden eklenmesini ve cilt lezyonu yanıtlarının izlenmesini içermektedir. Glutenin tekrardan verilmesinden sonra semptomların iyileşmesi veya kötüleşmesi dermatitis herpetiformis tanısını destekleyebilir.
Tedavi
Dermatitis herpetiformis için ana tedavi ömür boyu sıkı bir glutensiz diyettir. Bu, buğday, arpa, çavdar ve bunların türevleri dahil olmak üzere tüm gluten kaynaklarından kaçınmak anlamına gelir. Diyete uymak cilt lezyonlarını kontrol etmeye ve daha fazla hasarı önlemeye yardımcı olabilir.
Dapson, dermatitis herpetiformis (DH) tedavisi sırasında semptomların yönetilmesine ve deri lezyonlarını kontrol altına almaya yardımcı olmak için kullanılır.
Glutensiz diyete ek olarak, semptomları hafifletmek ve enflamasyonu azaltmak için ilaçlar reçete edilebilir. Sülfapiridin, bazı kortikosteroidler ve immünosupresanlar gibi diğer ilaçlar da bazı durumlarda kullanılabilir. Bir dermatolog veya gastroenterolog tarafından düzenli takip ziyaretleri, tedaviye yanıtı izlemek, komplikasyonları yönetmek ve genel sağlık ve refahı sağlamak için önemlidir.
If you want to spend more time about gluten enteropathy, have a look at this website
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Ortostatik hipotansiyon (OH), ayakta dururken veya dik bir pozisyondayken kan basıncında ani bir düşüşle karakterize bir durumdur. Genellikle ortostatik hipotansiyon olarak adlandırılır. Ortostatik hipotansiyon, vücut dik pozisyonda kan basıncını ve beyne ve diğer hayati organlara kan akışını korumak için yeterli telafiyi yapmadığında ortaya çıkar.
Anlamı:
Ortostatik hipotansiyon, ayakta durduktan veya ayakta durduktan sonraki 3 dakika içinde kan basıncında bir düşüş (genellikle sistolik basınçta en az 20 mmHg’lik bir düşüş veya diyastolik basınçta 10 mmHg’lik bir düşüş) olarak tanımlanır.
Belirtiler ve bulgular:
Baş dönmesi veya sersemlik: Ortostatik hipotansiyonu olan kişiler ayakta dururken genellikle baş dönmesi, sersemlik ve hatta baygınlık hissederler.
Bulanık görme: Göze giden kan akışının azalması nedeniyle görme bulanıklaşabilir veya kararabilir.
Bayılma: Ciddi vakalarda ortostatik hipotansiyon, geçici bir bilinç kaybı olan bayılmaya neden olabilir.
Zayıflık ve yorgunluk: Bazı kişiler, özellikle uzun süre ayakta durduktan sonra genel halsizlik ve yorgunluk yaşayabilir.
Bilişsel Bozukluk: Ortostatik hipotansiyon bilişsel işlevi bozar ve zayıf konsantrasyon, kafa karışıklığı ve hafıza sorunlarına yol açabilir.
Teşhis
Ortostatik hipotansiyonun teşhisi tıbbi öykü, fizik muayene ve spesifik testlerin bir kombinasyonunu gerektirir. Şunları içerebilir:
Kan basıncı ölçümü: Kan basıncı hasta yatarken ve ayakta durduktan sonra ölçülür ve duruş değişikliklerine bağlı olarak kan basıncındaki düşüş değerlendirilir.
Semptom değerlendirmesi: Kişilere ayağa kalkarken baş dönmesi veya bayılma gibi semptomlar ve bunların ne zaman başladığı sorulur.
Aktif ayakta durma veya eğik masa testi: Bazı durumlarda hastalar resmi bir eğik masa muayenesinden de geçebilir. Bu test hastayı dik bir pozisyona yerleştirir ve kan basıncı ve kalp atış hızındaki değişiklikleri izler.
Kan testi: Anemi, diyabet ve adrenal yetmezlik gibi ortostatik hipotansiyona neden olabilecek altta yatan bozuklukları kontrol etmek için kan testleri yapılabilir.
Otonom fonksiyonel test: Duruştaki değişikliklere otonom sinir sistemi yanıtını değerlendirmek için eğik masa testi ve otonom fonksiyon testleri gibi özel testler yapılabilir
Ortostatik hipotansiyon tedavisi semptomları hafifletmeyi ve altta yatan nedeni tedavi etmeyi amaçlar.
Yaklaşımlar şunları içerir:
Yaşam tarzı değişiklikleri: Sıvı ve tuz alımının artırılması, varis çorabı giyilmesi ve uzun süre ayakta durmaktan kaçınılması kan basıncının düzenlenmesine yardımcı olabilir.
Dozajlama: Ortostatik hipotansiyonu tedavi etmek için çeşitli ilaçlar reçete edilebilir. Bunlar arasında fludrokortizon (sıvı retansiyonunu artırmak için kullanılır), midodrin (kan damarlarını daraltmak için kullanılır) veya kan basıncını düzenlemeye yardımcı olan diğer ilaçlar yer alabilir.
Altta yatan hastalıkların yönetimi: Ortostatik hipotansiyon diyabet veya Parkinson hastalığı gibi altta yatan bir bozukluktan kaynaklanıyorsa, altta yatan bozukluğun tedavisi semptomları iyileştirmek için önemlidir.
Fiziksel önlemler: Ayakta durmadan önce bacak bacak üstüne atmak, çömelmek ve eğilmek gibi teknikler beyne giden kan akışını artırabilir ve semptomları azaltmaya yardımcı olabilir.
Ayırıcı tanı:
Ortostatik hipotansiyon, baş dönmesi ve bayılmanın diğer nedenlerinden ayırt edilmelidir. Ayırıcı tanılar arasında aritmiler, vazovagal senkop, bazı ilaçlar, iç kulak bozuklukları veya diğer otonom sinir sistemi bozuklukları yer alabilir.
Ortostatik hipotansiyonun erken tespiti ve uygun tedavisi semptomları en aza indirmek, düşmeleri önlemek ve yaşam kalitesini artırmak için önemlidir. Nörolog gibi bir tıp uzmanıyla konuşun
Doğru bir teşhis ve bireysel ihtiyaçlarınıza göre uyarlanmış uygun bir tedavi planı için bir doktora veya kardiyoloğa görünmeniz önerilir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Opsoklonus-miyoklonus sendromu (OMS), hızlı, kontrolsüz göz hareketleri (opsoklonus) ve ani, kısa kas spazmlarının (miyoklonus) bir kombinasyonu ile karakterize nadir bir nörolojik bozukluktur. Hastalık öncelikle çocukları etkiler, ancak yetişkinlerde de ortaya çıkabilir. OMS genellikle altta yatan nöroblastom ile ilişkilidir ve bazen paraneoplastik bir sendrom olarak sınıflandırılır.
Anlamı:
Opsoklonus, “dans eden gözler” görüntüsü veren, birden fazla yönde istemsiz, hızlı ve düzensiz göz hareketidir. Miyoklonus ise çeşitli kas gruplarını etkileyebilen ani, kısa süreli bir kas kasılması veya spazmıdır. OMS’de hem opsoklonus hem de miyoklonus bir arada bulunur ve karakteristik semptom ve bulgulara neden olur. Belirti ve bulgular:
Opsoklonus: Ana belirti, farklı yönlerde spontan ve kaotik göz hareketlerinin varlığıdır. Bu göz hareketleri genellikle istemsizdir ve istemli veya refleksif hareketlerle şiddetlenebilir.
Miyoklonus: Miyoklonus ani, kısa kas kasılmaları veya spazmları ile karakterizedir. Çeşitli kas gruplarını etkileyebilir ve uzuvların, gövdenin ve yüzün kontrolsüz hareketlerine yol açabilir. Miyoklonus simetri ve asimetriye sahiptir.
Ataksi: OMS’ye genellikle kas koordinasyonu ve denge eksikliği anlamına gelen ataksi eşlik eder. Çocuklar dengesiz bir yürüyüşe veya koordinasyon güçlüğüne sahip olabilir.
Davranış değişikliği: OMS’li çocuklar sinirlilik, ruh hali değişimleri ve uyku bozuklukları sergileyebilir.
Konuşma Bozukluğu ve Bilişsel Bozukluk: OMS’li bazı kişilerde konuşma ve dil bozuklukları ve hatta bilişsel bozukluklar görülür.
Teşhis
Opsoklonus-miyoklonus sendromunun teşhisi, aşağıdakileri içeren kapsamlı bir değerlendirme gerektirir:
Tıbbi geçmiş: Başlangıcı, seyri ve ilişkili semptomları değerlendirmek için kapsamlı bir tıbbi öykü alınır.
Klinik muayene: Nörolojik muayeneler göz hareketlerini, kas tonusunu, koordinasyonu ve refleksleri değerlendirmeye odaklanır.
Klinik muayene: Enfeksiyon, otoimmün belirteçler veya altta yatan nöroblastom kanıtlarını kontrol etmek için kan ve idrar testleri yapılır. 4. Nörogörüntüleme: Beyin ve omurganın manyetik rezonans görüntüleme (MRI) gibi görüntüleme testleri, yapısal anormallikleri ve nöroblastom varlığını tespit etmek için yapılabilir.
Nörofizyolojik çalışmalar: Beyin elektriksel aktivitesini ve kas tepkisini değerlendirmek için elektroensefalografi (EEG) ve elektromiyografi (EMG) yapılabilir.
Nöroblastomun değerlendirilmesi: OMS’den şüpheleniliyorsa, ilişkili nöroblastomu tanımlamak için görüntüleme çalışmaları ve belirli biyobelirteç testleri gibi daha ileri araştırmalar yapılır.
Süreç:
Opsoklonik miyoklonik sendromun tedavisi, altta yatan nedeni tedavi etmeye, semptomları hafifletmeye ve destekleyici bakım sağlamaya odaklanan disiplinler arası bir yaklaşım gerektirir. Tedavi seçenekleri şunları içerir:
İmmünoterapi: Bağışıklık sistemini modüle etmek ve semptomları azaltmak için kortikosteroidler, intravenöz immünoglobulin (IVIG) veya immünosupresanlar kullanılabilir.
Tümör tedavisi: Altta yatan bir nöroblastom tespit edilirse, cerrahi, kemoterapi veya radyasyon tedavisi gibi uygun tedavi başlatılır.
Semptomatik tedavi: Benzodiazepinler, antiseizure ilaçlar veya belirli semptomları (miyoklonus gibi) hedefleyen diğer ilaçlar gibi ilaçlar semptomları hafifletmek için reçete edilebilir.
Destekleyici bakım: Hareket bozukluklarını tedavi etmek, genel işlevi iyileştirmek ve gelişimi teşvik etmek için fiziksel, mesleki ve konuşma terapisi kullanılabilir.
Opsoklonus miyoklonus tanısı
Bu sendrom, benzer semptomlarla ortaya çıkan diğer hastalıklardan ayırt edilmelidir. Ayırıcı tanı esansiyel tremor, Tourette sendromu veya diğer miyoklonus formları gibi diğer hareket bozukluklarını içerebilir. Ensefalit, epilepsi ve metabolik bozukluklar gibi nörolojik bozukluklar da göz önünde bulundurulmalı ve ayrıntılı olarak değerlendirilmelidir.
Opsoklonik-miyoklonik sendromun karmaşıklığı ve altta yatan nöroblastom ile olası ilişkisi göz önüne alındığında, uygun tedavi ve yönetim için hızlı ve doğru tanı esastır. OMS’li hastaların kapsamlı bakımı ve desteği için nörologlar, onkologlar ve diğer sağlık profesyonelleri arasında işbirliğine dayalı bir yaklaşım esastır.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Olive pontocerebellar atrofi (OPCA), öncelikle beyincik, beyin sapı ve merkezi sinir sisteminin diğer kısımlarını etkileyen ilerleyici bir nörodejeneratif hastalıktır. Belirli bölgelerdeki nöronların dejenerasyonu ve kaybı ile karakterize olup hareket bozukluklarına ve diğer nörolojik semptomlara yol açar. OPCA, koordinasyon ve dengeyi etkileyen bir grup genetik bozukluk olan spinoserebellar ataksinin bir türüdür.
Anlamı:
Olive pontocerebellar atrofi, zeytin köprüsü, pons ve serebellumdaki nöronların dejenerasyonunu ifade eder. Başlangıç yaşına ve altta yatan genetik mutasyonlara göre farklı tiplere ayrılır. OPCA’nın ana özelliği ilerleyici ataksidir. Bu, düzensiz hareketlere ve denge zorluklarına neden olan kas koordinasyonu ve kontrol eksikliğini ifade eder.
Belirtiler ve bulgular:
Ataksi: OPCA’nın karakteristik bir belirtisi, hem ince hem de kaba motor becerilerini etkileyebilen ataksidir. Hastalar dengesiz bir yürüyüş, titreme, koordinasyon ve denge sorunları ve sarsıntılı veya kesin olmayan hareketler yaşayabilir.
Dizartri: Konuşma bozukluğu, ses kalitesinde değişiklikler ve anlaşılabilirlik kaybı gibi konuşma bozuklukları OPCA’da yaygındır.
Anormal göz hareketleri: Hastalarda aşağıdaki gibi göz hareketi anormallikleri gelişebilir: B. Nistagmus (istemsiz göz hareketleri), kompulsif intrüzyon (hızlı kontrolsüz göz hareketleri) veya okülopati (yanlış göz hareketleri).
Parkinsonizm Özellikleri: OPCA’lı bazı kişiler sertlik, bradikinezi (yavaş hareketler) ve postüral instabilite gibi Parkinson benzeri semptomlar sergiler.
Diğer nörolojik semptomlar: OPCA ayrıca otonomik disfonksiyon, bilişsel bozukluk, uyku bozukluğu ve psikiyatrik semptomlar olarak da ortaya çıkabilir.
Teşhis
Olivopontoserebellar atrofi tanısı klinik muayene, aile öyküsü değerlendirmesi, nörolojik muayene ve özel testlerin bir kombinasyonunu içerir. Bu, beyin atrofisinin derecesini değerlendirmek ve diğer olası nedenleri ekarte etmek için manyetik rezonans görüntüleme (MRI) gibi beyin görüntüleme testlerini içerebilir. Genetik testler, OPCA’nın birçok farklı genetik formu olduğu için OPCA ile ilişkili spesifik genetik mutasyonların belirlenmesine yardımcı olabilir.
Süreç
Şu anda OPCA için bir tedavi yoktur ve tedavi semptomları hafifletmeye ve hastanın yaşam kalitesini artırmaya odaklanmıştır. Tedavi yaklaşımları şunları içerir:
Fizyoterapi: Egzersiz ve rehabilitasyon programları denge, koordinasyon ve hareketliliği geliştirmeyi amaçlamaktadır.
Yardımlar: Yürüteç ve baston gibi hareketlilik yardımcıları, hastaların dengelerini ve bağımsızlıklarını korumalarına yardımcı olur. 3. Dozajlama: Titreme, spastisite ve psikotik semptomlar gibi belirli semptomları tedavi etmek için ilaçlar reçete edilebilir.
Destekleyici bakım: Psikolojik destek, konuşma terapisi ve mesleki terapi duygusal sağlığın, iletişim zorluklarının ve günlük yaşam aktivitelerinin yönetilmesine yardımcı olabilir.
Ayırıcı tanı:
Doğru tedavi için OPCA’yı diğer ataksilerden ve nörodejeneratif hastalıklardan ayırt etmek önemlidir. Benzer semptomlarla ortaya çıkabilen hastalıklar arasında diğer spinoserebellar ataksi formları, çoklu sistem atrofisi (MSA), Parkinson hastalığı ve bazı nadir genetik bozukluklar yer alır. Kapsamlı bir laboratuvar, görüntüleme, genetik ve özel nörolojik muayene, doğru bir ayırıcı tanı koymaya yardımcı olacaktır.
OPCA ilerleyici olduğundan, semptom kontrolünü optimize etmek, işlevi en üst düzeye çıkarmak ve bu rahatsızlığa sahip bireylere yeterli desteği sağlamak için erken tanı ve devam eden tedavi kritik öneme sahiptir. Nörologlar, fizyoterapistler ve diğer sağlık profesyonellerini içeren multidisipliner bir yaklaşım, olivopontoserebellar atrofisi olan hastaların kapsamlı bakımı için gereklidir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
İnfantil epileptik ensefalopati veya infantil inhibitör patlama epilepsi sendromu olarak da bilinen Otahara sendromu, genellikle yaşamın ilk birkaç ayı içinde başlayan nadir ve şiddetli bir epilepsi türüdür. Sendrom adını, 1970’lerde sendromu ilk kez rapor eden Japon nörolog Profesör Ohtahara’dan almıştır.
Anlamı:
Otahara sendromu sık nöbetler, anormal elektroensefalogramlar (EEG veya EEG) ve gelişimsel gecikme ile karakterizedir. Nörogelişimi olumsuz etkileyen bir grup ciddi epileptik bozukluk olan epileptik ensefalopati olarak sınıflandırılır.
Belirtiler ve bulgular:
Nöbetler: Otahara sendromlu bebekler, genellikle günde birkaç kez olmak üzere sık sık nöbet geçirir. Bu nöbetler genellikle inatçıdır, bu da ilaçların çok iyi çalışmadığı anlamına gelir.
Anormal EEG: EEG izleme, ‘bastırma patlamaları’ olarak bilinen karakteristik bir model gösterir. Bu patern, yüksek genlikli ani yükselmeler veya keskin dalga patlamalarının ardından düz veya düze yakın EEG aktivitesi dönemlerinden oluşur.
Gelişimsel gecikme: Otahara sendromlu çocuklarda gelişimsel gecikme ve zihinsel engellilik vardır. Oturma, emekleme ve yürüme gibi gelişimsel kilometre taşlarına ulaşmak zor olabilir.
Nörolojik anormallikler: Bebeklerde hipertoni (artmış kas tonusu) veya hipotoni (azalmış kas tonusu) gibi anormal kas tonusu ve anormal refleksler olabilir.
Diğer ilgili özellikler: Otahara sendromlu bazı kişilerde görme sorunları, beslenme güçlükleri, solunum sorunları ve diğer nörolojik komplikasyonlar görülür.
Teşhis:
Otahara sendromunun teşhisi, klinik belirtilerin, EEG bulgularının kapsamlı bir şekilde değerlendirilmesini ve nöbetlerin ve gelişimsel gecikmenin diğer olası nedenlerinin dışlanmasını gerektirir. Genetik testler, bu sendromla ilişkili altta yatan genetik mutasyonu tanımlayabilir. Manyetik rezonans görüntüleme (MRG) gibi nörogörüntüleme çalışmaları beyin yapısının değerlendirilmesine ve diğer yapısal anormalliklerin elenmesine yardımcı olur.
Süreç:
Antiepileptik ilaçlar: Nöbetleri tedavi etmek ve kontrol altına almak için çeşitli antiseizür ilaçlar reçete edilebilir. Ancak bu ilaçların Otahara sendromunda etkinliği sınırlı olabilir.
Ketojenik Diyet: Bazı hastalar, bazı epileptik bozukluklarda nöbet sıklığını azalttığı gösterilen yüksek yağlı, düşük karbonhidratlı bir diyet olan ketojenik diyetten fayda görebilir.
Destekleyici bakım: Tedavi, ilişkili semptomları yönetmeyi ve komplikasyonları yönetmek için destekleyici bakım sağlamayı içerir. Bunlar, gelişimsel ilerlemeyi desteklemek ve yaşam kalitesini artırmak için fiziksel, mesleki ve konuşma terapisini içerebilir.
Ayırıcı tanı:
Doğru tedavi için Otahara sendromunu diğer epileptik ensefalopatilerden ve nöbet bozukluklarından ayırt etmek önemlidir. Benzer semptomlara sahip olabilecek bozukluklar arasında diğer erken başlangıçlı epileptik ensefalopatiler (West sendromu ve Dravet sendromu gibi), metabolik bozukluklar, yapısal beyin anormallikleri ve genetik sendromlar yer alır. Klinik özelliklerin, EEG paternlerinin, genetik testlerin ve nörogörüntülemenin dikkatli bir şekilde değerlendirilmesi, doğru bir ayırıcı tanı konulmasına yardımcı olur.
Otahara sendromunun karmaşıklığı göz önüne alındığında, nörologları, genetikçileri ve diğer tıp uzmanlarını içeren multidisipliner bir yaklaşım, bu rahatsızlığa sahip bireylere en uygun bakım ve desteği sağlamak için gereklidir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Oksipital nevralji, başın arkasında veya oksipital bölgede kronik ağrı ile karakterize nörolojik bir hastalıktır. Bu ağrı genellikle elektrik çarpması veya bıçaklanma hissine benzeyen keskin bir bıçak ağrısı olarak tanımlanır. Kafatasının tabanında bulunan ve kafa derisine kadar uzanan oksipital sinirin tahrişi veya iltihaplanmasından kaynaklanır.
Belirtiler ve bulgular:
Şiddetli baş ağrısı: Ana belirti, çoğunlukla başın bir tarafında lokalize olan tekrarlayan baş ağrılarıdır.
Gözlerin arkasında ağrı: Bazı kişiler başın arkasından etkilenen taraftaki göze doğru yayılan bir ağrı hisseder.
Işığa duyarlılık: Parlak ışıklar ve aşırı görsel uyarım ağrıyı artırabilir.
Kafa Derisi Hassasiyeti: Kafa derisi, özellikle oksipital sinirin geçtiği bölgelerde dokunmaya karşı hassas olabilir.
Boyun hareketlerinin neden olduğu ağrı: Belirli boyun hareketleri ve pozisyonları, B. başın geriye döndürülmesi veya eğilmesi, ağrıyı tetikleyebilir veya şiddetlendirebilir.
Teşhis:
Oksipital nevraljinin teşhisi, semptomların kapsamlı bir şekilde değerlendirilmesini ve bir doktor tarafından fizik muayene yapılmasını gerektirir. Doktor hastanın tıbbi geçmişini değerlendirecek, kapsamlı bir nörolojik muayene yapacak ve ağrının diğer olası nedenlerini ekarte etmek için tanısal testler yapacaktır. Bu testler, baş ve boyun yapılarını değerlendirmek için MRI ve BT taramaları gibi görüntüleme testlerini içerebilir.
Süreç:
İlaçlar: Ağrıyı hafifletmek ve enflamasyonu azaltmak için steroid olmayan anti-enflamatuar ilaçlar (NSAID’ler), kas gevşeticiler ve ağrı kesiciler reçete edilebilir.
Sinir bloğu: Örneğin, oksipital sinir tıkanıklığı, lokal anestezik enjeksiyonları gibi etkilenen siniri felç ederek geçici bir rahatlama sağlayabilir.
Fizyoterapi: Germe, duruş düzeltme ve manuel terapi gibi teknikler boyun hareketliliğini artırmaya ve sinir iltihabını azaltmaya yardımcı olabilir.
Sinir stimülasyonu: Etkilenen sinire elektriksel uyarılar veren bir cihazın implante edilmesini içeren oksipital sinir stimülasyonu, diğer tedavilere yanıt vermeyen kişiler için düşünülebilir.
Yaşam tarzı değişiklikleri: Tetikleyicilerden kaçınmak, stres seviyelerini yönetmek, gevşeme teknikleri uygulamak ve iyi duruşu korumak ağrının başlamasını önlemeye veya en aza indirmeye yardımcı olabilir.
Ayırıcı tanı:
Oksipital nevralji diğer durumlarla bazı benzerlikler gösterir ve baş ve boyun ağrısının diğer nedenlerinden ayırt edilmesi için kapsamlı bir muayene gerektirir. Benzer semptomlar gösterebilen hastalıklar arasında migren, servikojenik baş ağrıları, gerilim tipi baş ağrıları, trigeminal nevralji ve servikal omurga hastalığı yer alır. Bir sağlık uzmanı hastanın semptomlarını ve tıbbi geçmişini değerlendirecek ve oksipital nevraljiyi ayırt etmek ve doğru bir şekilde teşhis etmek için tanısal testler yapacaktır. Oksipital nevralji semptomları olan kişilerin doğru bir teşhis ve özel ihtiyaçları için uygun bir tedavi planı almak için bir doktora görünmeleri önemlidir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Tay-Sachs hastalığı, merkezi sinir sistemini etkileyen nadir bir genetik bozukluktur. Beyin ve omurilikteki sinir hücrelerinin ilerleyici yıkımı ile karakterizedir. Tay-Sachs hastalığına hekzosaminidaz A (Hex-A) adı verilen bir enzimdeki eksiklik neden olur ve bu da sinir hücrelerinde GM2 gangliosid adı verilen yağlı maddelerin birikmesine yol açar.
Anlamı:
Tay-Sachs hastalığı otozomal resesif bir hastalıktır ve hastalığın gelişmesi için her iki ebeveynin de genin mutasyona uğramış bir kopyasını taşıması gerekir. Çoğunlukla bebekleri ve küçük çocukları etkiler ve ciddi nörolojik hasara ve yaşam süresinin kısalmasına yol açar. Belirtiler ve bulgular:
Tay-Sachs hastalığının belirtileri genellikle bebeklik döneminde ortaya çıkar, ancak geç başlangıçlı Tay-Sachs hastalığı olarak adlandırılan daha az yaygın bir form ergenlik veya yetişkinlik döneminde ortaya çıkabilir. Belirtiler ve semptomlar şunlardır:
Nörogelişimsel gecikme: Tay-Sachs hastalığı olan bebekler yuvarlanma, oturma ve emekleme gibi gelişimsel kilometre taşlarına ulaşamayabilir. Azalmış kas tonusu (düşük kan basıncı) egzersiz kapasitesinde gecikmeye neden olabilir.
Retinada kiraz rengi lekeler: Tay-Sachs hastalığının karakteristik bir belirtisi, retinanın merkezinde göz muayenesi sırasında görülebilen kiraz kırmızısı bir yamanın varlığıdır. Bunun nedeni retina içinde gangliosid birikimidir.
İlerleyici egzersiz kapasitesi kaybı: Hastalık ilerledikçe, etkilenen bireyler aşağıdakiler de dahil olmak üzere önceden edinilmiş motor becerilerde kayıp yaşarlar: B. Emekleme, oturma ve yürüme yeteneği. Ayrıca kas güçsüzlüğü ve sertliği de gelişebilir. 4. Nöbetler: Tay-Sachs hastalığında infantil spazmlar ve miyoklonik ataklar da dahil olmak üzere nöbetler yaygındır.
Bilişsel ve entelektüel gerileme: Tay-Sachs hastalığı olan çocuklar, dil kaybı ve zeka geriliği de dahil olmak üzere ilerleyici bilişsel gerileme yaşarlar.
Yutma güçlüğü: Hastalık ilerledikçe yutma sorunları ortaya çıkabilir, bu da yetersiz beslenme ve kilo kaybına yol açabilir.
Teşhis
Tay-Sachs hastalığının teşhisi, aşağıdakiler de dahil olmak üzere birkaç adım gerektirir:
Genetik test: Genetik testler, Hex-A enziminin üretilmesinden sorumlu olan HEXA genindeki mutasyonların varlığını doğrulayabilir. Bu gendeki mutasyonlar Tay-Sachs hastalığına işaret eder.
Enzim analizi: Hex-A enzimatik aktivite seviyesini değerlendirmek için enzimatik bir test yapılabilir. Tay-Sachs hastalığı hastalarında Hex-A enzimatik aktivitesi ciddi şekilde eksiktir.
Doğum öncesi test: Tay-Sachs hastalığı öyküsü olan veya yüksek risk taşıyan ailelerde, gelişmekte olan fetüste gen mutasyonunun varlığını tespit etmek için doğum öncesi test yapılabilir.
Süreç:
Şu anda Tay-Sachs hastalığının tedavisi yoktur ve tedavi semptomları yönetmeye ve destekleyici bakım sağlamaya odaklanır. Tedavi seçenekleri şunları içerir:
Semptom yönetimi: Nöbetleri kontrol etmek, kas sertliği ve spastisite semptomlarını hafifletmek ve ortaya çıkabilecek diğer tıbbi sorunları tedavi etmek için ilaçlar reçete edilebilir.
Destekleyici bakım: Fizik tedavi, mesleki terapi, konuşma terapisi ve beslenme desteğini içeren multidisipliner bir yaklaşım semptomları azaltmaya, yaşam kalitesini artırmaya ve işlevi optimize etmeye yardımcı olabilir.
Palyatif bakım: Hastalık ilerledikçe, palyatif bakımın odak noktası kişinin ve ailesinin sağlığını sağlamak ve mümkün olan en iyi yaşam kalitesini sürdürmektir.
Ayırıcı tanı:
Tay-Sachs hastalığı diğer nörodejeneratif hastalıklarla bazı benzerlikler gösterir:
Sandhoff hastalığı: Sandhoff hastalığı, Hex-A ve Hex-B enzimlerindeki kusurların neden olduğu ilgili bir lizozomal depolama bozukluğudur. Belirtileri ve seyri Tay-Sachs hastalığınınkine benzerdir.
Niemann-Pick hastalığı tip A: Bu, aşağıdakilerle karakterize edilen başka bir lizozomal depolama bozukluğudur: Hücre içi lipid birikimi. Nörogelişimsel gerileme ve retinada kiraz kırmızısı lekeler gibi bazı klinik özellikleri Tay-Sachs hastalığı ile paylaşır.
Canavan hastalığı: Canavan hastalığı, beyindeki beyaz maddenin ilerleyici yıkımı ile karakterize nadir bir genetik bozukluktur. Belirtiler Tay-Sachs hastalığı ile örtüşebilir. Tay-Sachs hastalığının doğru teşhisi, uygun genetik tedavi için kritik öneme sahiptir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Perinöral kist olarak da adlandırılan Tarlov kisti, omurilik yakınındaki bir sinir kökünde gelişen sıvı dolu bir boşluktur. Bu kistler genellikle omurganın sakral (lomber) kısmında meydana gelir, ancak diğer bölgelerde de oluşabilirler. Tarlov kistleri genellikle iyi huylu olmalarına rağmen semptomlara neden olabilir ve bazen tıbbi müdahale gerektirebilir.
Anlamı
Tarlov kisti, bir sinir kökü, özellikle de spinal sinir kökü ile omuriliğin koruyucu örtüsünün (dura mater) birleştiği yerde oluşan beyin omurilik sıvısı dolu bir kesedir. Bu kistler genellikle asemptomatiktir, ancak komşu sinir yapılarını büyütebilir veya sıkıştırarak ağrıya ve diğer nörolojik semptomlara neden olabilirler. Belirtiler ve bulgular:
Tarlov kistinin varlığı mutlaka semptomlara neden olmaz. Bununla birlikte, semptomlar ortaya çıktığında, bunlar aşağıdaki gibi semptomları içerebilir:
Ağrı: En yaygın belirti bel, sakrum, kalça veya pelvik bölgede kronik, lokalize ağrıdır. Ağrı donuk, ağrılı veya bıçak saplanır tarzdadır ve bacaklara ve cinsel organlara yayılabilir.
Sinirle ilgili semptomlar: Büyük veya semptomatik bir Tarlov kisti yakındaki sinir köklerini sıkıştırabilir ve siyatik (siyatik sinire yayılan ağrı), bacaklarda uyuşma veya karıncalanma, halsizlik ve yürüme güçlüğü dahil olmak üzere çeşitli nörolojik semptomlara neden olabilir. Ben de var.
Mesane ve bağırsak fonksiyon bozukluğu: Bazı durumlarda, sakral bölgedeki Tarlov kistleri sadece bağırsak rahatsızlığına değil, aynı zamanda aciliyet, aciliyet ve inkontinansa da neden olabilir.
Teşhis
Tarlov kistlerinin teşhisi klinik değerlendirme ve görüntüleme çalışmalarının bir kombinasyonunu gerektirir. Teşhis yöntemleri şunları içerir:
Tıbbi öykü ve fizik muayene: Doktorlar kişinin semptomlarını ve tıbbi geçmişini değerlendirir ve sinir fonksiyonunu değerlendirmek için fiziksel muayene yapar.
Görüntüleme: Manyetik rezonans görüntüleme (MRG), Tarov kistlerini görüntülemek ve değerlendirmek için en güvenilir görüntüleme yöntemidir. MR, kistin yerini, boyutunu ve kapsamını belirlemeye ve ilişkili sinir sıkışmasını veya hasarını tanımlamaya yardımcı olabilir.
Süreç
Tarlov kistinin tedavisi semptomları hafifletmeyi ve yaşam kalitesini artırmayı amaçlamaktadır. Tedavi seçenekleri şunları içerir:
Konservatif yönetim: Bir Tarlov kisti küçük ve asemptomatikse, konservatif yönetim önerilebilir. Bu, kistteki değişikliklerin izlenmesini ve semptomların ağrı kesiciler, fizik tedavi ve yaşam tarzı değişiklikleri ile tedavi edilmesini içerir.
Müdahale Prosedürü: Konservatif önlemlere yanıt vermeyen semptomatik Tarlov kistleri olan kişiler için girişimsel tedavi düşünülebilir. Bu prosedürler arasında kist aspirasyonu (kist sıvısının boşaltılması) veya kist fenestrasyonu (sıvının boşalmasını sağlamak için kistin spinal sıvı boşluğuna bağlanması) yer alır.
Cerrahi müdahale: Nadiren, semptomlar günlük yaşamı ciddi şekilde etkileyecek kadar şiddetliyse, kistin cerrahi olarak çıkarılması düşünülebilir. Bununla birlikte, Tarlov kistlerinin cerrahi tedavisi tartışmalıdır ve genellikle seçilmiş vakalar için ayrılmıştır.
Ayırıcı tanı
Tarlov kisti ile ilişkili semptomlar diğer omurga hastalıklarının semptomlarına benzeyebilir. Ayırıcı tanılar şunları içerir:
Disk herniasyonu: Lomber omurgada fıtıklaşmış bir disk yakındaki sinirlere baskı yapabilir ve bel ve bacaklarda ağrı, uyuşma ve güçsüzlük gibi semptomlara neden olabilir.
Spinal stenoz: Spinal stenoz, omurilik ve sinir köklerini sıkıştırarak bacaklarda ağrı, uyuşma ve güçsüzlüğe neden olabilen spinal kanalın daralmasıdır.
Diğer kistik lezyonlar: Araknoid kistler ve sinovyal kistler gibi diğer kistik lezyonlar omurga bölgesinde gelişebilir ve Tarlov kistlerine benzer semptomlara neden olabilir.
Ürat tanısı ve Tarlov kistlerinin uygun tedavisi, omurga hastalıkları konusunda deneyimli bir doktor tarafından kapsamlı bir değerlendirme yapılmasını gerektirir. Tedavi yaklaşımları, kistin boyutu ve yeri, semptomların şiddeti ve günlük yaşam üzerindeki etkisi göz önünde bulundurularak bireyselleştirilmelidir. Semptomları izlemek ve tedavi planlarını gerektiği gibi ayarlamak için düzenli takip ziyaretleri şarttır.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Tardif diskinezi (TD), yüz, dil, uzuvlar ve gövdenin istemsiz ve tekrarlayan hareketleriyle karakterize nörolojik bir bozukluktur. Bazı ilaçların, özellikle antipsikotiklerin uzun süreli kullanımının bir yan etkisidir ve tedavisi zor olabilir.
Anlamı:
Tardif diskinezi, özellikle antipsikotikler olmak üzere dopamin reseptör blokerlerine uzun süre maruz kalmanın bir sonucu olarak ortaya çıkan bir hareket bozukluğudur. Bu ilaçlar yaygın olarak şizofreni, bipolar bozukluk ve bazı nörolojik bozukluklar gibi psikiyatrik bozuklukları tedavi etmek için kullanılır.
Belirtiler ve bulgular:
Tardif diskinezinin semptom ve bulguları şiddete ve sunuma göre değişir, ancak genellikle istemsiz hareketleri içerir ve iki kategoriye ayrılabilir:
Orofasiyal diskineziler: Dudak şapırdatma, dil çıkarma, yüz buruşturma, kırıştırma veya ısırma gibi yüzün veya ağzın istemsiz hareketleri.
Uzuv ve gövde diskinezileri: Uzuvlarda ve gövdede tekrarlayan sarsıntı, bükülme ve kıvranma hareketleri gibi istemsiz hareketler. Diğer belirti ve semptomlar şunları içerir:
Kore hareketleri: Uzuvların, ellerin ve parmakların hızlı, sarsıntılı, düzensiz hareketleri.
Atetoid hareket: Özellikle parmaklarda yavaş, kıvranma, bükülme hareketleri.
Distoni: Anormal duruşlara ve tekrarlayan hareketlere neden olan sürekli kas kasılmaları.
Akatizi: Öznel huzursuzluk ve hareket etme dürtüsü.
Kene: Kısa, tekrarlayan, ani bir hareket veya gürültü.
Teşhis koymak:
Tardif diskinezi teşhisi, aşağıdakileri içeren kapsamlı bir değerlendirme gerektirir:
Geçmiş ve İlaçların Gözden Geçirilmesi: Doktorlar, özellikle antipsikotikler ve TD ile ilişkili diğer ilaçlar olmak üzere mevcut ve geçmiş ilaç kullanımı da dahil olmak üzere hastanın tıbbi geçmişini gözden geçirecektir. 2. Fiziksel muayene: Anormal istemsiz hareketleri gözlemlemek ve değerlendirmek için kapsamlı bir muayene yapılır.
Derecelendirme Ölçeği: Tardif diskinezilerin şiddetini ve etkisini ölçmek için Anormal İstemsiz Hareket Ölçeği (AIMS) gibi çeşitli standardize edilmiş derecelendirme ölçekleri kullanılır. süreç: Tardif diskinezileri tedavi etmeye yönelik temel yaklaşımlar, semptomları azaltmayı veya kontrol etmeyi ve muhtemelen tedavi rejimlerini değiştirmeyi amaçlayan stratejilerin bir kombinasyonunu içerir. Tedavi seçenekleri şunları içerir:
İlaç hazırlama: Bazı durumlarda, bir doktorun rehberliğinde rahatsız edici ilacın ayarlanması veya durdurulması semptomları hafifletebilir. Ancak bu karar, bireyin altta yatan tıbbi koşulları ve etkili tedavi ihtiyacı ışığında dikkatle değerlendirilmelidir.
İlaç değişikliği: Bazı durumlarda, mevcut antipsikotiğin tardif diskinezi riski daha düşük olan başka bir antipsikotik ile değiştirilmesi düşünülebilir. Bu, bir doktor gözetiminde yapılmalıdır.
Semptomları hafifletmek için ilaçlar: Tardif diskinezi ile ilişkili hareketleri kontrol etmek için tetrabenazin gibi bazı ilaçlar reçete edilebilir. Bu ilaçlar beyindeki dopamini parçalayarak çalışır.
Botulinum toksin enjeksiyonu: Bazı durumlarda, belirli kasları hedef almak ve istemsiz hareketlerin şiddetini geçici olarak azaltmak için botulinum toksini (Botoks) enjeksiyonları kullanılabilir.
Destekleyici bakım: Fiziksel, mesleki ve konuşma terapisi, tardif diskinezi ile ilişkili işlevsel zorlukların yönetilmesine ve yaşam kalitesinin iyileştirilmesine yardımcı olabilir. Ayırıcı tanı: Tardif diskinezi, aşağıdakiler gibi benzer semptomlara sahip diğer hareket bozukluklarından ayırt edilmelidir:
Ayırıcı tanı:
Tardif diskinezi, benzer semptomlara sahip diğer hareket bozukluklarından ayırt edilmelidir, örn:
Parkinson hastalığı: Parkinson hastalığı titreme, sertlik ve bradikinezi (yavaş hareket) ile karakterize ilerleyici bir nörolojik bozukluktur. Özellikle uzun süre antipsikotik kullanan kişilerde tardif diskinezi ile karıştırılabilir.
İlaçla ilişkili diğer hareket bozuklukları: Bazı antiemetikler ve duygudurum dengeleyiciler gibi diğer ilaçlar tardif diskineziye benzer hareket bozukluklarına neden olabilir.
Diğer birincil hareket bozuklukları: Huntington hastalığı, Tourette sendromu ve diğer distoniler gibi bozukluklar tardif diskinezilerle örtüşen semptomlara sahip olabilir.
Tardif diskinezilerin doğru teşhis ve tedavisi, hareket bozuklukları konusunda deneyimli bir doktor tarafından kapsamlı bir değerlendirme yapılmasını gerektirir. Tedavi stratejileri, semptomların şiddeti, tedavi edilen altta yatan hastalık ve bireyin genel sağlığı dikkate alınarak bireyselleştirilmelidir. Tedavi yanıtını değerlendirmek ve tedavi rejimlerini gerektiği gibi ayarlamak için düzenli izleme ve takip şarttır.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Nörosifiliz olarak da adlandırılan dorsal enfeksiyon, öncelikle merkezi sinir sistemini etkileyen sifiliz enfeksiyonunun geç bir belirtisidir. Omuriliğin dorsal kolonunun dejenerasyonu ile karakterizedir ve çeşitli nörolojik semptom ve bulgulara neden olur.
Anlamı:
Sırt ağrısı, tedavi edilmemiş veya yetersiz tedavi edilmiş sifiliz enfeksiyonunun neden olduğu ilerleyici bir nöropatidir. Omurilikte iltihaplanma ve dejenerasyona neden olan Treponema pallidum bakterisinin uzun süreli hasarının sonucudur.
Belirtiler ve bulgular:
Sırt ağrısı birden fazla vücut sistemini etkileyen çeşitli semptom ve bulgularla ortaya çıkabilir. En yaygın özellikler şunlardır:
Duyu bozukluğu: Omuriliğin dorsal kolonunun dejenerasyonu duyusal eksikliklere neden olur. Bu, propriyosepsiyon (duruş hissi) kaybı ve titreşim aşırı duyarlılığı olarak ortaya çıkabilir. Hastalar denge ve koordinasyonda zorluk yaşayabilir.
Bıçak saplanır gibi ağrı: Sırt ağrısına genellikle özellikle bacaklarda ve ayaklarda keskin, bıçak saplanır gibi ağrılar eşlik eder. Ağrı aniden ortaya çıkabilir, şiddetlenebilir ve geceleri daha da kötüleşebilir. Mide Krizi:
Sırt tüberkülozu olan kişilerde mide krizi adı verilen şiddetli karın ağrısı, kusma ve ishal belirtileri görülebilir.
Mesane disfonksiyonu: Dorsal sekmeler mesane disfonksiyonuna neden olarak idrar kaçırma, idrar retansiyonu veya eksik işeme ile sonuçlanabilir.
Cinsel işlev bozukluğu: Erektil disfonksiyon ve libido kaybı erkeklerde yaygındır, ancak kadınlar orgazma ulaşmakta zorluk çekebilir.
Argyll Robertson öğrencileri: Argyll-Robertson gözbebeği olarak bilinen spesifik bir oküler bulgu mevcut olabilir. Bu durum, yakın bir nesneye odaklanırken göz bebeğinin daralması, ancak ışıktaki değişikliklere karşı duyarsızlık ile karakterizedir.
Teşhis:
Dorsal nodüllerin teşhisi laboratuvar testleri, tıbbi geçmiş ve bazı laboratuvar testlerinin bir kombinasyonunu gerektirir:
Laboratuvar testleri: Duyusal bozukluklar, disrefleksi ve diğer nörolojik belirtileri değerlendirmek için kapsamlı bir nörolojik muayene yapılır.
Tıbbi geçmiş ve seroloji: Sifiliz enfeksiyonu olasılığı da dahil olmak üzere ayrıntılı bir tıbbi öykü alınır. Frengi antikorlarının varlığını belirlemek için zührevi hastalıklar laboratuvarı (VDRL) testi ve treponemal antikor testi gibi kan testleri kullanılır.
Beyin omurilik sıvısı (BOS) analizi: Artmış beyaz kan hücresi sayısı, protein seviyeleri ve Treponemal sifiliz antikorlarının varlığı gibi nörosifiliz belirtileri açısından BOS’u analiz etmek için bir spinal musluk yapılır.
Nörogörüntüleme: Beyin ve omuriliğin manyetik rezonans görüntülemesi (MRI) gibi görüntüleme testleri, yapısal anormallikleri aramak ve nörolojik semptomların diğer nedenlerini dışlamak için yapılabilir.
Süreç:
Dorsal tüberkülozun tedavisi, altta yatan sifiliz enfeksiyonunun uygun antibiyotik tedavisi, genellikle yüksek doz intravenöz penisilin ile tedavisini içerir. Antibiyotik tedavisinin süresi ve türü sifiliz enfeksiyonunun evresine ve şiddetine bağlıdır. Bazı durumlarda, nöropatik ağrıyı azaltmak için ek ağrı yönetimi stratejileri de kullanılabilir.
Ayırıcı tanı:
Sırt ağrısı, benzer nörolojik semptomlara neden olabilen diğer hastalıklardan ayırt edilmelidir, örneğin:
Diyabetik nöropati: Diyabetik nöropatiye, özellikle alt ekstremitelerde duyusal eksiklikler ve nöropatik ağrı da eşlik edebilir.
Multipl Skleroz: Multipl skleroz, duyusal bozukluklar ve mesane disfonksiyonu da dahil olmak üzere benzer nörolojik semptom ve bulgulara neden olabilen otoimmün bir hastalıktır.
B12 Vitamini eksikliği: B12 vitamini eksikliği nörolojik ve duyusal eksikliklere yol açabilir.
Dorsal ısırıkları benzer semptomların diğer olası nedenlerinden ayırt etmek ve uygun tedaviyi sağlamak için ayrıntılı bir tıbbi öykü, fizik muayene, klinik muayene ve görüntüleme çalışmalarını içeren kapsamlı bir değerlendirme şarttır. Sifiliz enfeksiyonunun erken teşhisi ve hızlı tedavisi, dorsal tüberkülozun ilerlemesini önlemek ve uzun vadeli nörolojik hasarı en aza indirmek için kritik öneme sahiptir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
İstismarcı kafa travması veya sarsılmış darbe sendromu olarak da bilinen Sarsılmış Yenidoğan Sendromu (SBS), yenidoğan veya küçük bir çocuğun şiddetli bir şekilde sarsıldığı ciddi bir çocuk istismarı şeklidir. Ciddi beyin hasarı ve diğer bedensel zararlarla sonuçlanabilen kasıtlı veya kasıtsız sarsma ile ayırt edilir.
Sarsılmış Bebek Sendromu, bir bebeğin ya da küçük çocuğun şiddetle sarsılması sonucu oluşan travmatik bir beyin hasarıdır. Sarsıntı, beynin kafatası içinde dönmesine neden olarak kan damarlarının yırtılmasına, kanamaya ve beyin büyümesine yol açabilir.
Sarsılmış Bebek Sendromu Belirtileri ve Göstergeleri:
Sarsılmış Bebek Sendromunun semptomları ve göstergeleri hasarın ciddiyetine bağlı olarak değişir, ancak ortak yönler şunları içerebilir:
Sinirlilik, yorgunluk, farkındalıkta azalma, kasılmalar veya koma, hafif ila şiddetli nörolojik semptomlara örnektir.
Solunum problemleri: Sarsıntı solunum sistemi hasarına neden olarak solunum problemleri, apne (kısa süreli nefes durması) veya solunum yetmezliği ile sonuçlanabilir.
Fiziksel göstergeler arasında baş, yüz veya boyunda morarma veya şişme, retinal kanamalar (gözlerin arkasında kanama), kaburga veya diğer kemik kırıkları ve diğer fiziksel istismar biçimlerinin kanıtları yer alır.
Davranış değişiklikleri: Çocuklarda aşırı ağlama, yeme güçlükleri veya uyku düzeninde değişiklikler gibi davranış değişiklikleri görülebilir.
Gelişimsel gecikmeler: Sarsılmış Bebek Sendromunun uzun vadeli sonuçları arasında gelişimsel gecikmeler, zihinsel engellilik, öğrenme güçlükleri veya davranış sorunları yer alabilir.
Teşhis:
Sarsılmış Bebek Sendromu, aşağıdakileri içeren tam bir tıbbi değerlendirmeyi içerir:
Tıp uzmanları, çocuğun semptomları ve olası travmatik olaylar hakkında bilgi edinmek için bakıcılar veya tanıklarla görüşecektir.
Fiziksel muayene: Beyin yaralanmaları, kırıklar veya diğer fiziksel bulgular gibi travma belirtilerini aramak için kapsamlı bir muayene yapılır.
Bilgisayarlı tomografi (BT) taramaları veya manyetik rezonans görüntüleme (MRI) gibi görüntüleme incelemeleri beyin lezyonlarını, kanamayı veya şişmeyi tespit etmek için gereklidir.
Bir göz doktoru, şiddetli kafa travması vakalarında yaygın olan retina kanamalarını aramak için genişlemiş göz muayenesi yapabilir.
Enfeksiyonlar veya metabolik anormallikler gibi semptomların diğer potansiyel nedenlerini ekarte etmek için kan testleri yapılabilir.
Tedavi:
Sarsılmış Bebek Sendromu tedavisi çocuğun durumunu stabilize etmeye, acil zorlukları gidermeye ve destekleyici bakım vermeye çalışır.
Tedavi yöntemleri şunları içerebilir: yaralanmaların ciddiyetine bağlı olarak:
Çocuğun sağlığını stabilize etmek ve potansiyel olarak hayatı tehdit eden yaralanmaları veya sonuçları yönetmek için acil tıbbi yardım şarttır.
Ciddi beyin hasarı veya kanama varsa, beyin üzerindeki baskıyı azaltmak veya kafatası kırıklarını düzeltmek için cerrahi müdahale gerekebilir.
Fizik tedavi, mesleki terapi ve konuşma terapisi gibi rehabilitasyon hizmetleri, gelişimsel gecikmeleri, motor bozuklukları veya bilişsel anormallikleri ele almak için sıklıkla kullanılır.
Uzun vadeli yönetim, çocuk ve ailesi için duygusal desteğin yanı sıra sürekli tıbbi izleme gerektirir.
Ayırıcı Tanı:
Sarsılmış Bebek Sendromu şüphesi olan bir çocuğu tararken, semptomların aşağıdakileri içerebilecek ek olası açıklamaları dikkate alınmalıdır:
Kaza sonucu travma: Kazara düşme veya diğer kasıtsız yaralanmaların semptomları zaman zaman istismara bağlı kafa travmasınınkilerle eşleşebilse de, öykü ve fizik muayene bulguları ikisini birbirinden ayırmaya yardımcı olabilir.
Kan bozuklukları veya metabolik bozukluklar gibi bazı tıbbi hastalıklar Sarsılmış Bebek Sendromuna benzer semptomlara neden olabilir. Bu durumlar laboratuvar testleri ve ileri tanı muayeneleri ile tespit edilebilir.
Sarsılmış Bebek Sendromu tanısını değerlendirmek ve doğrulamak, çocuğa uygun bakımı sağlamak ve aileye destek olmak için çocuk doktorları, nörologlar, göz doktorları ve sosyal hizmet uzmanları dahil olmak üzere sağlık uzmanlarından oluşan bir ekip tarafından kapsamlı bir değerlendirme yapılması gerekir. Çocuğun güvenliğini ve refahını sağlamak için şüpheli çocuk istismarı vakalarını ilgili makamlara bildirmek çok önemlidir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
De Morsier sendromu veya optik sinir hipoplazisi olarak da bilinen SOD, optik sinirlerin, hipofiz bezinin ve çevre organların gelişimini bozan nadir bir doğumsal hastalıktır. Optik sinir anomalileri, septum pellusidumun (orta hat beyin yapısı) az gelişmişliği veya yokluğu ve hormonal eksiklikler bu hastalığı karakterize eder.
Septo-Optik Displazi, fetal gelişim sırasında ortaya çıkan ve optik sinirlerin ve ilgili beyin bölgelerinin uygunsuz gelişimiyle sonuçlanan bir bozukluktur. Kesin nedeni genellikle bilinmemektedir, ancak genetik ve çevresel faktörler söz konusu olabilir.
Belirtiler ve İşaretler:
Septo-Optik Displazi belirtileri ve bulguları kişiden kişiye değişir, ancak ortak özellikler şunları içerebilir:
Görme sorunları: Optik sinir hipoplazisi veya gelişmemiş optik sinirler, görme keskinliğinde azalma, görme alanı anormallikleri veya körlük gibi küçük ila ciddi görme bozukluklarına neden olabilir.
Septo-Optik Displazi, hipotalamik ve hipofiz bezi anormallikleri ile ilişkilidir ve hormon eksikliğine neden olur. Bu durum büyüme hormonu eksikliği, tiroid hormonu eksikliği ve adrenal yetmezlikle sonuçlanabilir; bu da bodur büyüme, gecikmiş ergenlik ve diğer endokrin bozukluklara yol açabilir.
SOD’lu çocuklarda motor beceriler, dil gelişimi ve bilişsel yetenek gibi gelişimsel kilometre taşlarının tamamlanmasında gecikmeler meydana gelebilir.
Öğrenme güçlükleri, dikkat eksikliği hiperaktivite bozukluğu (DEHB) ve bilişsel bozukluklar görülebilir.
Nöbetler: Diğer nörolojik hastalıklarda olduğu kadar yaygın olmasa da SOD’li bazı kişilerde nöbetler görülebilir.
Septo-Optik Displazi, aşağıdakileri içeren eksiksiz bir tıbbi değerlendirme ile teşhis edilir:
Kapsamlı bir tıbbi öykü ve fizik muayene yapılır. Optik sinir bozukluklarını ve görme fonksiyonunu belirlemek için göz muayenesi ve görme keskinliği testleri kullanılır. Nörogörüntüleme: Beynin manyetik rezonans görüntülemesi (MRI) optik sinir hipoplazisi ve septum pellusidum yokluğu gibi yapısal anormallikleri tespit edebilir. Hormon seviyelerini izlemek ve endokrin eksikliklerini teşhis etmek için kan testleri yapılabilir.
Septo-Optik Displazi tedavisi semptom yönetimi ve gelişimsel sonuçları optimize etmeye odaklanır. Mevcut tedavi seçenekleri arasında şunlar yer almaktadır:
Görme işlevini iyileştirmek için düzeltici lensler, görsel yardımcılar ve az görme terapisi reçete edilebilir.
Hormon replasman tedavisi: Hormon eksiklikleri, büyüme hormonu, tiroid hormonu ve kortikosteroidleri içerebilen hormon replasman tedavisi ile tedavi edilir.
Erken müdahale hizmetleri: Fizik tedavi, mesleki terapi, konuşma terapisi ve eğitim desteği, gelişimsel gecikmeler ve öğrenme sorunlarına yardımcı olabilir.
Nöbet yönetimi: Nöbetler meydana gelirse antiepileptik ilaçlar sağlanabilir.
Sürekli gözetim: Büyümeyi, hormon seviyelerini ve genel gelişimi değerlendirmek için düzenli tıbbi kontroller yaptırmak çok önemlidir.
Ayırıcı Tanı:
Septo-Optik Displaziyi taklit edebilecek diğer bozukluklar şunlardır:
Optik sinir atrofisi: Optik sinir dejenerasyonu veya hasarı ile karakterize, görme kaybıyla sonuçlanan bir bozukluk.
İzole hormon eksiklikleri: Bazı hormon eksiklikleri görme siniri tutulumu olmaksızın ortaya çıkabilir.
Orta hat beyin kusurları: Korpus kallozum agenezisi gibi diğer orta hat beyin anomalileri de benzer semptomlar gösterebilir.
Septo-Optik Displaziyi diğer bozukluklardan ayırt etmek ve etkilenen hastalara uygun bakım ve desteği yönlendirmek için pediatristler, göz doktorları, endokrinologlar ve nörologlar dahil olmak üzere multidisipliner bir uzman ekibi tarafından kapsamlı bir değerlendirme yapılması gerekir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Şizensefali, serebral hemisferlerde anormal yarıklar veya derin, sıvı dolu yarıklar ile ayırt edilen nadir bir konjenital beyin deformitesidir. Bu yarıklar gri madde ile kaplıdır ve beyin yüzeyinden ventriküllere kadar uzanır.
Şizensefali, serebral hemisferlerin düzgün bir şekilde oluşmadığı fetal gelişim sırasında ortaya çıkan bir durumdur. Spesifik etiyolojisi bilinmemekle birlikte genetik ve çevresel faktörlerle ilişkili olduğu düşünülmektedir.
Belirtiler ve Bulgular:
Şizensefali belirtileri ve bulguları yarıkların boyutuna, konumuna ve kapsamına göre değişir. Ortak özellikler şunları içerebilir:
Şizensefalili çocuklarda oturma, emekleme veya yürüme gibi gelişimsel kilometre taşlarına ulaşmada gecikmeler yaygındır. Entelektüel bozukluk son derece yaygındır.
Nöbetler şizensefalinin yaygın bir belirtisidir ve şiddeti ve sıklığı değişebilir.
Motor bozukluklar arasında güçsüzlük, spastisite ve motor koordinasyon ve kontrolde zorluk yer alır.
Öğrenme güçlükleri, dikkat eksikliği hiperaktivite bozukluğu (DEHB) ve davranış sorunları da mümkündür.
Konuşma ve dil gelişiminde gecikmeler: Şizensefalili çocuklar konuşma ve dil gelişiminde gecikmeler yaşayabilir.
Görme alanı kusurları veya körlüğün yanı sıra işitme kaybı da dahil olmak üzere görsel anormallikler görülebilir.
Hidrosefali: Bazı durumlarda, sıvı dolu yarıklar beyin omurilik sıvısının geçişini engelleyerek hidrosefali (beyinde aşırı sıvı birikmesi) ile sonuçlanabilir.
Teşhis Şizensefali genellikle beynin manyetik rezonans görüntülemesi (MRG) gibi görüntüleme incelemeleri yoluyla teşhis edilir, bu da serebral hemisferlerdeki karakteristik yarıkları tespit edebilir. Hastalıkla bağlantılı altta yatan genetik anormallikleri ekarte etmek için genetik testler de yapılabilir.
Tedavi:
Şizensefali tedavisi semptomları yönetmeyi ve yaşam kalitesini artırmayı amaçlar. Genellikle aşağıdakileri içeren multidisipliner bir yaklaşım gerektirir:
Nöbetlerin kontrol altına alınması antiepileptik ilaçların kullanılmasını gerektirebilir. Fiziksel ve mesleki terapi, motor fonksiyon, hareketlilik ve günlük yaşam aktivitelerine yardımcı olabilir. Konuşma ve dil tedavisi: Konuşma ve dil terapisi iletişim becerilerinizi geliştirmenize yardımcı olabilir. Hareket ve bağımsızlığı geliştirmek için tekerlekli sandalye veya diş teli gibi hareket yardımcıları reçete edilebilir. Destekleyici bakım: Hidrosefali gibi ilgili sorunları kontrol altına almak için düzenli tıbbi değerlendirmelerin ve müdahalelerin yapılması çok önemlidir.
Ayırıcı Tanı:
Şizensefali, benzer semptomlara sahip diğer beyin anormallikleri veya bozuklukları gibi görünebilir veya bunlarla karıştırılabilir:
Porensefali beyinde sıvı dolu deliklerle karakterize edilir, ancak altta yatan etiyoloji farklıdır. Holoprosensefali, yetersiz serebral hemisfer ayrımı ile tanımlanan bir grup beyin anomalisidir. Aicardi sendromu, diğer özelliklerinin yanı sıra korpus kallozum eksikliği ile karakterize nadir bir genetik durumdur.
Şizensefaliyi diğer benzer hastalıklardan ayırmak ve uygun tedaviyi önermek için görüntüleme taramaları ve genetik testler de dahil olmak üzere tam bir nörolog muayenesi gereklidir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Bentall ameliyatı, aort kökü anevrizmalarını ve aort kapak hastalığını, özellikle de çıkan aort söz konusu olduğunda, tedavi etmek için kullanılan cerrahi bir prosedürdür. Prosedür adını 1960’larda geliştiren İngiliz kardiyotorasik cerrah Hugh Bentall’dan almıştır.
Bentall ameliyatında hasarlı aort kapağı ve aort kökü kompozit bir greft ile değiştirilir. Kompozit greft, sentetik tübüler greft ve mekanik veya doku kapakçıktan oluşur. Bu prosedür genellikle genel anestezi altında gerçekleştirilir ve aşağıdaki adımları içerir:
Meme kesisi: Kalbe ve aorta erişmek için göğüste orta hattan bir kesi yapılır. Aort kapağı ve aort kökünün çıkarılması: Hasarlı aort kapağı ve aortun genişlemiş veya anevrizmal kısmı çıkarılır.
Kompozit Greft Yerleştirilmesi: Aort kökünün yerine genellikle Dacron veya benzer bir malzemeden yapılmış sentetik bir tüp greft yerleştirilir. Greft hastanın anatomisine uyum sağlar ve yerine dikilir.
Kapak replasmanı: Greft üzerine mekanik veya biyolojik bir kapak takılır. Kapak tipinin seçimi hastanın yaşı, genel sağlık durumu ve cerrahın tercihi dahil olmak üzere birçok faktöre bağlıdır. Koroner reattachment: Kalbi besleyen koroner arterler, miyokarda yeterli kan akışını sağlamak için kompozit grefte dikkatlice yeniden bağlanır.
Kapatma: Göğüs kesisi kapatılır ve hasta iyileşme alanına transfer edilir.
Bentall ameliyatı hem hasarlı aort kapağını hem de hastalıklı aort kökünü etkili bir şekilde değiştirerek kan akışını iyileştirir ve aort anevrizmalarıyla ilişkili daha fazla komplikasyonu önler. Bu büyük bir açık kalp ameliyatıdır ve ameliyat sonrası izleme ve iyileşme için hastanede kalmayı gerektirir. Bu prosedürü geçiren hastaların genellikle ömür boyu gözlemlenmesi gerekir ve kullanılan kapak tipine bağlı olarak kan pıhtılaşmasını önlemek için antikoagülan veya antiplatelet ilaçlar almaları gerekebilir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Kırılgan kemik hastalığı olarak da bilinen Osteogenezis imperfekta (OI), kırılmaya eğilimli kırılgan kemiklerle karakterize genetik bir bozukluktur. Öncelikle kemiklerin sağlamlığından ve yapısından sorumlu ana protein olan kolajen üretimindeki bir kusurdan kaynaklanır. OI’nin şiddeti hafiften ağıra kadar değişebilir. Osteogenezis imperfektanın tanımı, belirti ve bulguları, teşhisi, tedavisi ve ayırıcı tanısına ilişkin ayrıntılı bir açıklamayı burada bulabilirsiniz:
Tanım:
Osteogenezis imperfekta, kolajen üretiminde bir kusur ile karakterize, kırılmaya eğilimli kırılgan kemiklerle sonuçlanan genetik bir bozukluktur. Kolajen, kemik gücü ve yapısı için gerekli bir proteindir. OI, ilgili spesifik genetik mutasyona bağlı olarak otozomal dominant veya resesif bir şekilde kalıtılabilir. Şiddet ve ilişkili özelliklere göre değişen çeşitli OI türleri vardır.
Belirtiler ve Bulgular:
Osteogenezis imperfekta belirtileri ve bulguları, durumun türüne ve ciddiyetine bağlı olarak büyük ölçüde değişebilir. Ortak özellikler şunları içerebilir:
Sık kırıklar: OI’nin ayırt edici semptomu, genellikle minimal travma ile veya görünürde bir neden olmadan meydana gelen tekrarlayan kırıklardır. Kırıklar, hastalığın ciddiyetine bağlı olarak doğum sırasında, bebeklik döneminde veya yaşamın ilerleyen dönemlerinde ortaya çıkabilir.
Kemik deformiteleri: OI, uzun kemiklerin eğilmesi, omurganın anormal eğriliği (skolyoz) ve vertebral kompresyon kırıkları nedeniyle boy kısalması dahil olmak üzere kemik deformitelerine neden olabilir.
Mavi skleralar: Skleraların inceliği ve şeffaflığı nedeniyle göz akları mavi veya grimsi bir renk tonuna sahip olabilir.
İşitme kaybı: OI, kulaktaki anormal kemik oluşumu nedeniyle bazı bireylerde işitme kaybına yol açabilir.
Diş anormallikleri: Diş minesinin anormal gelişimi nedeniyle dişler renksiz, normalden küçük veya çürüklere eğilimli olabilir.
Eklem gevşekliği: Eklem hipermobilitesi veya gevşekliği mevcut olabilir, bu da eklem instabilitesine ve dislokasyon riskinin artmasına neden olur.
Solunum problemleri: Ağır OI vakalarında, göğüs deformiteleri akciğer fonksiyonlarını etkileyebilir ve solunum güçlüklerine yol açabilir.
Teşhis
Osteogenezis imperfekta tanısı klinik değerlendirme, tıbbi öykü, fizik muayene ve tanısal testlerin bir kombinasyonunu içerir. Aşağıdaki yaklaşımlar yaygın olarak kullanılmaktadır:
Tıbbi öykü ve fizik muayene: Doktor semptomları, tıbbi geçmişi, aile geçmişini değerlendirecek ve kemik deformiteleri veya mavi skleralar gibi OI belirtilerini aramak için kapsamlı bir fizik muayene yapacaktır.
Genetik test: Genetik testler, durumla ilişkili spesifik genetik mutasyonları tanımlayarak OI tanısını doğrulayabilir. OI’nin türünü ve kalıtım modelini belirlemeye yardımcı olabilir.
X ışınları: Röntgenler kemik yoğunluğunu, yapısını ve kırık veya deformite belirtilerini değerlendirmek için kullanılır. OI tanısını desteklemek için değerli bilgiler sağlayabilirler.
Kolajen analizi: Bazı durumlarda, kolajen üretimini ve yapısını analiz etmek için bir cilt biyopsisi yapılabilir. Bu test OI tanısını doğrulamaya yardımcı olabilir.
Tedavi
Osteogenezis imperfekta tedavisi semptomları yönetmeye, kırıkları önlemeye ve genel kemik sağlığını geliştirmeye odaklanır. Tedavi seçenekleri şunları içerebilir:
Kırık yönetimi: Kırıkların hızlı ve uygun tedavisi çok önemlidir. Bu, alçı veya atellerle immobilizasyonu, kırıkların cerrahi fiksasyonunu veya diş teli veya tekerlekli sandalye gibi destekleyici cihazların kullanımını içerebilir.
İlaçlar: Kemik yoğunluğunu artırmak ve kırık riskini azaltmak için pamidronat veya zoledronik asit gibi bifosfonatlar reçete edilebilir.
Fizik tedavi: Fizik tedavi kas gücünü, eklem stabilitesini ve hareketliliği artırmaya yardımcı olabilir. Güvenli hareketi teşvik etmek için egzersizler, esneme ve teknikler içerebilir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Schilder hastalığı, merkezi sinir sistemindeki (MSS) sinir liflerinin koruyucu örtüsü olan miyelin kılıfına zarar veren nadir bir nörolojik rahatsızlıktır. Miyelin parçalanması ve kaybı ile karakterize olan ve nörolojik bozukluğa neden olan bir multipl skleroz (MS) türüdür.
Schilder hastalığı, beynin hem beyaz hem de gri maddesini etkileyen geniş demiyelinizan lezyonların varlığı ile ayırt edilir. Schilder hastalığının spesifik kökeni bilinmemektedir, ancak bağışıklık sisteminin yanlış bir şekilde miyeline saldırdığı ve zarar verdiği bir otoimmün bileşen içerdiği düşünülmektedir.
Belirtiler ve Bulgular:
Schilder hastalığı semptom ve bulguları demiyelinizasyonun yeri ve yoğunluğuna göre değişir. Ortak özellikler şunları içerebilir:
Motor güçsüzlük, koordinasyon kaybı, spastisite, denge ve yürüme güçlükleri nörolojik bozukluklara örnektir.
Schilder hastalığı hafıza sorunları, dikkat ve odaklanma sorunları, davranış ve kişilik bozuklukları gibi bilişsel eksikliklere neden olabilir.
Nöbetler: Bazı kişilerde doğası ve sıklığı değişebilen nöbetler görülebilir.
Schilder hastalığı bulanık veya çift görme, görme alanı kaybı ve hatta körlük gibi görme zorluklarına neden olabilir.
Migren de dahil olmak üzere baş ağrıları bazı durumlarda mümkündür.
Schilder hastalığının nadir görülmesi ve diğer hastalıklarla benzerlik göstermesi nedeniyle teşhis edilmesi zordur.
Genellikle aşağıdakileri içeren kapsamlı bir muayene gerektirir:
Fiziksel muayene ve tıbbi geçmiş
Motor ve duyusal işlevleri, koordinasyonu ve refleksleri değerlendirmek için nörolojik muayene yapılır.
Beyin görüntüleme: Manyetik rezonans görüntüleme (MRG) beyindeki önemli demiyelinizan lezyonları tespit etmek için kritik öneme sahiptir. Ancak, benzer lezyonlar başka hastalıklarda da görülebilir ve bu da tanıyı zorlaştırır.
Spinal tap (lomber ponksiyon): Beyin omurilik sıvısı (BOS) analizi, enflamatuar bir sürecin belirtilerini aramak için yapılabilir.
Tedavi:
Schilder hastalığının kesin bir tedavisi olmadığından, tedavi semptom yönetimine ve hastalığın ilerlemesini geciktirmeye odaklanır. Olasılıklar arasında şunlar vardır:
Yüksek dozda kortikosteroidler: Bu tedaviler enflamasyonu ve immünolojik yanıtı azaltmaya yardımcı olabilir.
İmmünomodülatör tedavi: Multipl sklerozda, bağışıklık sistemini etkilemek için immünosupresanlar veya hastalık modifiye edici ilaçlar gibi ilaçlar kullanılabilir.
Fizik tedavi, mesleki terapi ve konuşma terapisi gibi rehabilitasyon terapileri, belirli semptomların yönetilmesine ve yaşam kalitesinin artırılmasına yardımcı olabilir.
Ayırıcı Tanı:
Birbiriyle örtüşen belirtiler nedeniyle Schilder hastalığını diğer bozukluklardan ayırt etmek zor olabilir. Ayırıcı tanılar aşağıdakileri içerebilir:
Multipl skleroz (MS): Schilder hastalığı MS’in bir türüdür ve iki hastalık arasında bazı benzerlikler vardır. Öte yandan Schilder hastalığı daha büyük ve daha yaygın lezyonlarla karakterizedir.
Akut yayılmış ensefalomiyelit (ADEM): ADEM, genellikle bir enfeksiyon veya aşıdan sonra ortaya çıkan bir tür akut inflamatuar demiyelinizan hastalıktır. Sıklıkla daha yaygın inflamasyon ve beyaz madde tutulumu ile kendini gösterir.
Diğer demiyelinizan hastalıklar: Nöromiyelitis optika (NMO), Balo’nun konsantrik sklerozu ve diğer olağandışı demiyelinizan sendromlar dikkate alınmayı gerektirebilir.
Schilder hastalığını diğer benzer hastalıklardan ayırmak ve tedaviyi yönlendirmek için görüntüleme incelemeleri ve laboratuvar testleri de dahil olmak üzere tam bir nörolog muayenesi gereklidir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
GM2 gangliosidoz tip 2 olarak da bilinen Sandhoff hastalığı, lizozomal depolama bozuklukları hastalık kategorisine ait nadir bir genetik hastalıktır. Otozomal resesif bir hastalıktır, yani bir kişi beta-heksozaminidaz adı verilen spesifik bir enzim üreten HEXB geninin iki kusurlu kopyasını miras alır.
Sandhoff hastalığı, beyinde ve diğer organlarda GM2 gangliosid olarak bilinen yağlı bir maddenin birikmesi ile ayırt edilir. İşlevsel bir beta-heksozaminidaz enziminin yokluğu bu kimyasalın birikmesine neden olarak merkezi sinir sistemine kademeli olarak zarar verir.
Belirtiler ve Bulgular:
Sandhoff hastalığı belirtileri tipik olarak bebeklik veya erken çocukluk döneminde ortaya çıkar ve zamanla kötüleşir. Belirti ve semptomlar arasında şunlar yer alır:
Gelişimde gecikmeler ve gerileme: Bebekler oturma, emekleme veya yürüme gibi gelişimsel kilometre taşlarına ulaşmada gecikmeler yaşayabilir. Ayrıca daha önce öğrendikleri becerilerde de kayıp yaşayabilirler.
Motor sorunları ve kas güçsüzlüğü: Sandhoff hastalığı olan çocuklar koordinasyon, kas zayıflığı ve hareketlerini kontrol etmede zorluk yaşayabilir.
Nöbetler: Nöbetler Sandhoff hastalığının yaygın bir belirtisidir ve şiddeti ve şekli değişebilir. Görme ve işitme sorunları: İlerleyici görme ve işitme kaybı meydana gelebilir.
Zihinsel engellilik: Sandhoff hastalığı olan çocuklar genellikle kademeli bir zihinsel engele sahiptir.
Retinada karakteristik kiraz kırmızısı leke: Göz muayenesinde retinada belirgin bir kiraz kırmızısı leke görülebilir, bu da hastalığın tanımlayıcı bir özelliğidir.
Hepatomegali (karaciğer büyümesi) ve splenomegali (dalak büyümesi) de olasılıklar arasındadır.
Solunum sorunları: Hastalık ilerledikçe solunum sorunları ortaya çıkabilir.
Sandhoff hastalığı klinik değerlendirme, genetik testler ve laboratuvar incelemelerinin bir kombinasyonu kullanılarak teşhis edilir. HEXB genindeki mutasyonlar genetik testler yoluyla tespit edilerek tanı doğrulanabilir. Ayrıca, kan, idrar veya doku örneklerinde yüksek GM2 gangliosid seviyeleri tanının doğrulanmasına yardımcı olabilir.
Tedavi:
Sandhoff hastalığının tedavisi olmadığından, tedavi semptom yönetimi ve destekleyici bakıma odaklanır. Mevcut tedavi seçenekleri arasında şunlar yer almaktadır:
Nöbetler, solunum güçlükleri ve diğer sonuçlar gibi semptomları tedavi etmek için ilaçlar önerilebilir.
Fiziksel ve mesleki terapi, hareketliliğinizi korumanıza ve yaşam kalitenizi artırmanıza yardımcı olabilir.
Destekleyici bakım: Organ fonksiyonlarını düzenli olarak kontrol etmek ve etkilenen kişilerin özel ihtiyaçlarını karşılamak çok önemlidir. Palyatif bakım semptom yönetimine ve yaşam kalitesine yardımcı olabilir.
Tanı Ayırıcı:
Sandhoff hastalığına benzer semptomlara neden olabilen diğer hastalıklar şunlardır:
Tay-Sachs hastalığı, beta-hekzosaminidaz A enziminin eksikliğinden kaynaklanan başka bir GM2 gangliosidoz türüdür.
Diğer lizozomal depo hastalıkları şunlardır: Gaucher hastalığı ve Niemann-Pick hastalığı gibi bozuklukların belirtileri örtüşebilir.
Sandhoff hastalığını diğer benzer bozukluklardan ayırt etmek için kapsamlı bir tıbbi muayene ve genetik testler gereklidir. Erken tanı, etkili yönetim ve destekleyici bakım için kritik öneme sahiptir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Melkersson-Rosenthal Sendromu (MRS), tekrarlayan veya kalıcı yüz ödemi, yüz felci ve fissürlü dil ile karakterize nadir görülen nörolojik bir durumdur. Klinik bir tanı olarak sınıflandırılır çünkü bozukluğu kesin olarak doğrulayacak belirli bir test mevcut değildir. İşte tanımı, semptom ve bulguları, teşhisi, tedavisi ve ayırıcı tanısına daha derinlemesine bir bakış:
Melkersson-Rosenthal Sendromu, orofasiyal ödem, fasiyal sinir felci (felç) ve çatlamış veya çatlamış dil gibi karakteristik üçlü semptomlarla ayırt edilen nadir bir nöro-enflamatuar hastalıktır. MRS’nin kökeni belli değildir, ancak anormal immünolojik yanıt ve kalıtsal faktörlerden kaynaklandığı düşünülmektedir.
Belirtiler ve Uyarı İşaretleri:
Melkersson-Rosenthal Sendromu aşağıdaki belirtilerle ayırt edilir:
En yaygın belirti dudakların, yanakların veya yüzün tekrarlayan veya inatçı şişmesidir. Şişlik asimetrik olabilir ve şiddeti değişebilir. Şişlik bazı durumlarda rahatsız edici veya hassas olabilir.
Yüz siniri felci: Bu durum yüz sinirinde felce veya zayıflamaya neden olarak yüzde sarkma, bir gözü kapatmada güçlük veya yüzdeki duyguların azalmasına yol açar. MRS sıklıkla fissürlü veya buruşuk bir dil ile ilişkilidir. Dilin yüzeyinde derin oluklar veya yarıklar olabilir.
Diğer olası semptomlar: MRS’li bazı kişilerde baş ağrısı, baş dönmesi, tinnitus (kulaklarda çınlama) veya işitme kaybı da gelişebilir.
Yüz ödeminin bulaşıcı veya otoimmün nedenlerini ekarte etmek ve genel sağlığı kontrol etmek için kan testleri yapılabilir.
Melkersson-Rosenthal Sendromu tedavisi, semptom yönetiminin yanı sıra nükslerin sıklığını ve şiddetini en aza indirmeye odaklanır.
Tedavi seçenekleri şunları içerebilir:
İlaçlar: Akut ataklar sırasında enflamasyonu azaltmak ve semptomları düzenlemek için steroid olmayan anti-enflamatuar ilaçlar (NSAID’ler), kortikosteroidler ve immünosupresif ilaçlar uygulanabilir.
Yüz felci vakalarında, yüz egzersizleri ve terapisi yüz kas gücünü ve işlevini iyileştirmeye yardımcı olabilir.
Kompresyon tedavisi: Sıkıştırma giysileri giymek veya yüze bandaj uygulamak şişlik ve rahatsızlığı en aza indirmeye yardımcı olabilir.
Tedavi, yüzdeki şişlik için ağrıyı hafifletme, kuru gözler için göz kayganlaştırıcıları veya konuşma bozuklukları için konuşma terapisi gibi semptomatik bakımı içerebilir.
Ayırıcı Tanı:
Melkersson-Rosenthal Sendromu ayırıcı tanısında aşağıdaki durumlar göz önünde bulundurulabilir:
Alerjik veya anjiyoödematöz reaksiyonlar: Akut veya kronik olsun, alerjik veya anjiyoödematöz reaksiyonlar yüzde şişliğe neden olabilir ancak MRS’nin tekrarlayan şişlik ve yüz siniri felci gibi ayırt edici özelliklerinden yoksundur.
Sarkoidoz, deri ve nörolojik sistem dahil olmak üzere birçok organı etkileyebilen granülomatöz bir hastalıktır. Belirgin semptomlar ve ileri tanısal testler MRS’den ayırt edilmesine yardımcı olabilir.
Nörosarkoidoz, merkezi sinir sistemini, yani kraniyal sinirleri etkileyen bir sarkoidoz türüdür. Yüz felci ile ortaya çıkabilmesine rağmen, sistemik tutulumun varlığı ve özel tanısal testler onu MRS’den ayırır.
Melkersson-Rosenthal Sendromunu teşhis etmek ve benzer semptom ve bulgulara sahip dier durumlardan ayrmak için bir dermatolog, nörolog veya az ve çene-yüz uzman tarafndan kapsaml bir deerlendirme gerekir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Genellikle Spinocerebellar Ataksi Tip 3 (SCA3) olarak bilinen genetik nörodejeneratif durum Machado-Joseph Hastalığı (MJD), öncelikle beyin ve omuriliği etkiler. Ataksi olarak bilinen kas koordinasyonu sorunlarının yanı sıra diğer nörolojik ve sistemik semptomların gelişmesiyle ayırt edilir. İşte tanımı, semptom ve bulguları, tanı, tedavi ve ayırıcı tanıya daha derinlemesine bir bakış:
Machado-Joseph Hastalığı (MJD), ATXN3 genindeki CAG tekrarlarının anormal amplifikasyonu ile karakterize kalıtsal bir durumdur. Büyümüş tekrar, beyin ve omuriliğin bazı bölgelerinde birikerek nöronal işlev bozukluğu ve dejenerasyona neden olan mutant bir protein olan ataksin-3’ün oluşmasına neden olur.
Belirtiler ve göstergeler: Machado-Joseph Hastalığı semptom ve göstergelerinin yoğunluğu ve gelişme yaşı değişiklik gösterir. Aşağıdakiler en yaygın özelliklerdir:
Ataksi: MJD’nin karakteristik özelliği koordinasyon ve denge ile ilgili ilerleyici sorunlardır. Genellikle yürüme, yazma ve ince motor becerileri gibi hareketler üzerinde etkisi vardır.
Dizartri ve disfaji dizartrinin iki belirtisidir. Bu görevlerde yer alan kasların tutulumu nedeniyle, konuşma bozukluğu veya yavaş konuşma (dizartri) ve yutma sorunları (disfaji) gibi konuşma sorunları ortaya çıkabilir.
Oftalmolojik belirtiler: Bazı kişilerde kontrol edilemeyen göz hareketleri (nistagmus), göz hareketlerini kontrol etmede zorluklar (oftalmopleji) ve göz koordinasyonunda azalma olabilir.
Spastisite ve kas sertliği: Spastisite ve artmış kas tonusu mevcut olabilir, bu da kas krampları, sertlik ve hareket etme güçlüğü ile sonuçlanabilir.
Periferik nöropati: Bu durum periferik sinirleri etkileyerek ekstremitelerde uyuşma, karıncalanma veya ağrı gibi duyusal anomalilere neden olur.
Parkinsonizm: Bazı kişilerde istirahat titremesi, bradikinezi (hareket yavaşlığı) ve sertlik gibi Parkinson semptomları gelişir.
Ruh hali ve bilişsel değişiklikler: İlerleyen aşamalarda, MJD depresyon ve anksiyete gibi ruh hali anormalliklerinin yanı sıra bilişsel bozukluk ve bunama ile de ilişkilendirilebilir.
Sistemik tutulum: MJD nadiren diğer sistemleri de etkileyerek kalp düzensizlikleri, solunum güçlükleri ve endokrin bozukluklara yol açabilir.
Teşhis Machado-Joseph Hastalığı klinik muayene, genetik testler ve görüntüleme testlerinin bir kombinasyonu kullanılarak teşhis edilir. Aşağıda temel tanı adımlarına örnekler verilmiştir:
MJD’ye özgü özellikleri değerlendirmek için tam bir tıbbi öykü, nörolojik muayene ve semptom ve bulguların değerlendirilmesi yapılır.
Genetik testler, ATXN3 genindeki CAG tekrarlarının anormal genişlemesini tanımlayarak MJD tanısını doğrulamaya yardımcı olur. Bu test için kan örnekleri kullanılabilir.
Görüntüleme çalışmaları: Beynin manyetik rezonans görüntülemesi (MRG), MJD’de beyincik ve beyin sapında atrofi (büzülme) gibi yaygın anormalliklerin belirlenmesine yardımcı olabilir.
Elektromiyografi (EMG) ve sinir iletim çalışmaları gibi elektrofizyolojik incelemeler, kaslardaki ve periferik sinirlerdeki elektriksel aktiviteyi ve sinir iletimini değerlendirerek diğer hastalıkların ekarte edilmesine ve periferik nöropatinin değerlendirilmesine yardımcı olabilir.
Şu anda Machado-Joseph Hastalığının tedavisi yoktur, bu nedenle tedavi semptom yönetimi ve destekleyici bakıma odaklanır. Tedavi seçenekleri şunları içerebilir:
Fiziksel ve mesleki terapi, kas koordinasyonu, denge ve hareketliliğin yönetimine ve iyileştirilmesine yardımcı olabilir.
Konuşma terapisi: Konuşma terapisi, konuşma bozuklukları ve yutma sorunlarına yardımcı olabilir.
İlaçlar: Spastisite için kas gevşeticiler veya ruh hali dengeleyiciler gibi belirli semptomları tedavi etmek için ilaçlar önerilebilir.
Yürüteç veya baston gibi yardımcı ekipmanlar kullanılarak hareketlilik ve denge geliştirilebilir.
Genetik danışmanlık: Etkilenen bireylere ve ailelerine, kalıtım modeli hakkında daha fazla bilgi edinmek ve aile planlaması kararlarına yardımcı olmak için genetik danışmanlık sunulmalıdır.
Ayırıcı Tanı:
Machado-Joseph Hastalığı tanısı konulurken aşağıdaki durumlar göz önünde bulundurulmalıdır:
Diğer spinoserebellar ataksi türleri (SCA): Çeşitli genetik anormalliklerin neden olduğu çeşitli diğer otozomal dominant spinoserebellar ataksiler vardır. SCA1, SCA2, SCA6, SCA7 ve SCA17 bunlara örnektir. Genetik testler bu bozukluklar arasında ayrım yapılmasına yardımcı olabilir.
Parkinson hastalığı: Parkinson hastalığının istirahat titremesi ve bradikinezi gibi bazı klinik semptomları MJD’ninkilere benzer. Bununla birlikte, ayırt edici ataksi ve genetik testler ikisini birbirinden ayırmaya yardımcı olabilir.
Friedreich ataksisi, kademeli koordinasyon eksikliklerine neden olan bir başka kalıtsal ataksi hastalığıdır. Bununla birlikte, ayrı bir genetik kökene sahiptir (FXN geninde bir sapma) ve farklı klinik semptomlarla kendini gösterir.
Machado-Joseph Hastalığının tanısını koymak ve benzer semptom ve bulgulara sahip diğer hastalıklardan ayırt etmek için, genetik testler ve görüntüleme tekniklerinin yanı sıra bir nörolog tarafından tam bir değerlendirme yapılması gerekir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Kluber-Bucy sendromu, beynin temporal loblarında, özellikle de amigdalada meydana gelen iki taraflı hasardan kaynaklanan benzersiz bir dizi semptomla karakterize nadir bir nörolojik bozukluktur. İlk olarak 1930’larda nörolog Heinrich Kluber ve Paul Bussy tarafından tanımlanmıştır.
Tanım
Kluber-Bucy sendromu, beynin temporal loblarının, özellikle de amigdalanın iki taraflı tutulumu veya işlev bozukluğundan kaynaklanan davranışsal bir sendromdur. En sık beyin hasarı, enfeksiyon veya temporal lobun dejeneratif hastalığı ile ilişkilidir.
Belirtiler ve Bulgular
Kluber-Bucy sendromu, aşağıdakiler de dahil olmak üzere bir dizi davranışsal ve nörolojik semptomla karakterizedir:
Oral Keşif ve Tıkınırcasına Yeme: Etkilenen bireyler kompulsif oral davranışlar sergileyebilir. B. Ağzınıza bir şeyler koyduğunuzda veya koyduğunuzda iştah artışı veya aşırı yeme yaşayabilirsiniz.
Görsel Agnozi: Görsel agnozi, görsel uyaranları algılayamama veya yorumlayamama anlamına gelir ve tanıdık nesneleri ve yüzleri tanımayı zorlaştırır.
İtaatkarlık ve azalmış anksiyete tepkisi: Kluber-Buchy sendromlu bireyler itaatkâr davranış, azalmış kaygı ve korku tepkileri ve azalmış saldırganlık sergileyebilir. Bu durum sosyal disinhibisyona ve uygunsuz etkileşimlere yol açabilir.
Hafıza bozukluğu: Özellikle yeni anıların oluşumunda hafıza bozukluğu görülebilir.
Apati ve duygusal değişiklikler: Apati, duygusal düzleşme ve duygusal tepkide azalma görülebilir, bu da duygusal ifade eksikliği ve uygunsuz duygusal tepkilerle sonuçlanabilir.
Hiperfaji: Hiperfaji, nesneleri ve ortamları ağız yoluyla keşfetmeye yönelik kompulsif bir eğilim anlamına gelir ve genellikle uygunsuz çiğneme ve emme davranışıyla sonuçlanır.
Görme bozukluğu: Görme bozukluğu halüsinasyonları, nesnelerin garip veya yabancı olarak algılanmasını ve duygusal yüz ifadelerinin tanınmasında bozulmayı içerebilir.
Teşhis
Kluber-Bucy sendromu, hastanın tıbbi geçmişinin, davranışsal semptomlarının ve nörogörüntüleme bulgularının kapsamlı bir şekilde değerlendirilmesini gerektirir. Teşhis prosedürleri şunları içerir:
Klinik Değerlendirme: Hiperseksüalite, sözel davranış, görsel agnozi ve duygusal değişiklikler gibi Kluber-Bucy sendromu ile ilişkili karakteristik semptomları tanımlamak için kapsamlı bir klinik değerlendirme yapılır.
Nörogörüntüleme: Manyetik rezonans görüntüleme (MRG) ve bilgisayarlı tomografi (BT) gibi görüntüleme testleri, temporal lobdaki, özellikle amigdaladaki yapısal anormallikleri ve lezyonları tespit etmek için kullanılır.
Nörodavranışsal testler: Bilişsel işlev, hafıza, görsel algı ve duygusal işlemeyi değerlendirmek için nörodavranışsal testler yapılabilir.
Laboratuvar Testleri: Enfeksiyonlar veya metabolik bozukluklar gibi semptomların diğer olası nedenlerini ekarte etmek için kan testleri ve diğer laboratuvar testleri istenebilir.
Tedavi: Tedavi: Aşağıdakiler için özel bir tedavi yoktur
Kluber-Bucy sendromu, bu nedenle tedavi öncelikle semptomları tedavi etmeye ve altta yatan nedeni ele almaya odaklanır. Tedavi seçenekleri şunları içerir:
Davranışsal Müdahaleler: Davranış terapisi ve çevresel modifikasyon, bireylerin uygunsuz davranışları yönetmesine ve yönlendirmesine yardımcı olur.
İlaçlar: Farmakolojik müdahalelerin hiperseksüalite, saldırganlık ve duygudurum bozuklukları gibi belirli durumları tedavi ettiği düşünülmektedir. Seçici serotonin geri alım inhibitörleri (SSRI’lar) ve antipsikotikler gibi ilaçlar bir psikiyatrist gözetiminde reçete edilebilir.
Destekleyici Bakım: Yapılandırılmış ve destekleyici bir ortamın sağlanması bireyin güvenliği ve refahı için kritik öneme sahiptir. Bu, hemşireler ve tıp uzmanları tarafından gözetim, rehberlik ve desteği içerebilir.
Ayırıcı tanı
Kluver-Bucy sendromu diğer nörolojik ve psikiyatrik bozukluklarla bazı benzerlikler gösterir ve alternatif nedenleri ekarte etmek için ayırıcı tanı önemlidir. Ayırıcı tanıda göz önünde bulundurulabilecek belirtiler şunlardır:
Temporal Lob Epilepsisi: Temporal lob nöbetleri, değişen davranışlar, bulanık görme ve hafıza sorunları gibi Kluber-Busy sendromuna benzer semptomlara neden olabilir.
Frontotemporal demans: Frontotemporal demansın belirli formları davranış değişiklikleri, duygusal hissizlik ve bozulmuş sosyal işlevsellik ile ilişkili olabilir.
Psikiyatrik Bozukluklar: Bipolar bozukluk, şizofreni ve dürtü kontrol bozuklukları gibi bazı psikiyatrik bozukluklar Kluber-Bucy sendromu ile örtüşebilir. Bu bozuklukların ayırt edilmesi psikiyatrik muayene ve değerlendirme gerektirir.
Kluber-Bucy sendromunun doğru bir şekilde teşhis edilmesi ve uygun tedavinin planlanması genellikle bir nörolog, bir nöropsikolog ve bir psikiyatristi içeren multidisipliner bir yaklaşımı gerektirir.
Merhaba arkadaşlar,
Ben, tibbiterimler.com’un kurucularından biriyim. Amacımız, tıbbi bilgiyi daha anlaşılır ve basit bir şekilde sunmaktır. İnsanlar genellikle düşündüklerinden daha karmaşık hale getiriyorlar. Aksine, sadece anlayan biri basitleştirebilir.
Web sitemizde size en iyi deneyimi sunmak için çerezleri kullanıyoruz. Bu siteyi kullanmaya devam ederseniz, bundan memnun olduğunuzu varsayacağız.Kabul ediyorumGizlilik-politikasi